de Smith Adam J, Walsh Kyle M, Hansen Helen M, Endicott Alyson A, Wiencke John K, Metayer Catherine, Wiemels Joseph L
Department of Epidemiology and Biostatistics, University of California San Francisco, San Francisco, California, United States of America.
Division of Neuroepidemiology, Department of Neurological Surgery, University of California San Francisco, San Francisco, California, United States of America.
PLoS One. 2015 Nov 17;10(11):e0143343. doi: 10.1371/journal.pone.0143343. eCollection 2015.
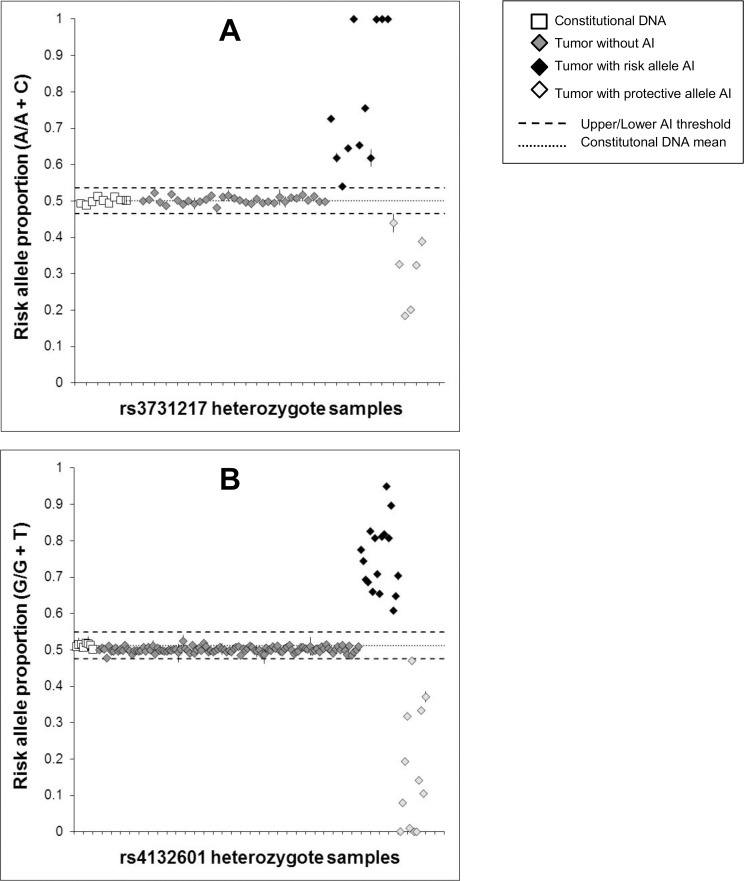
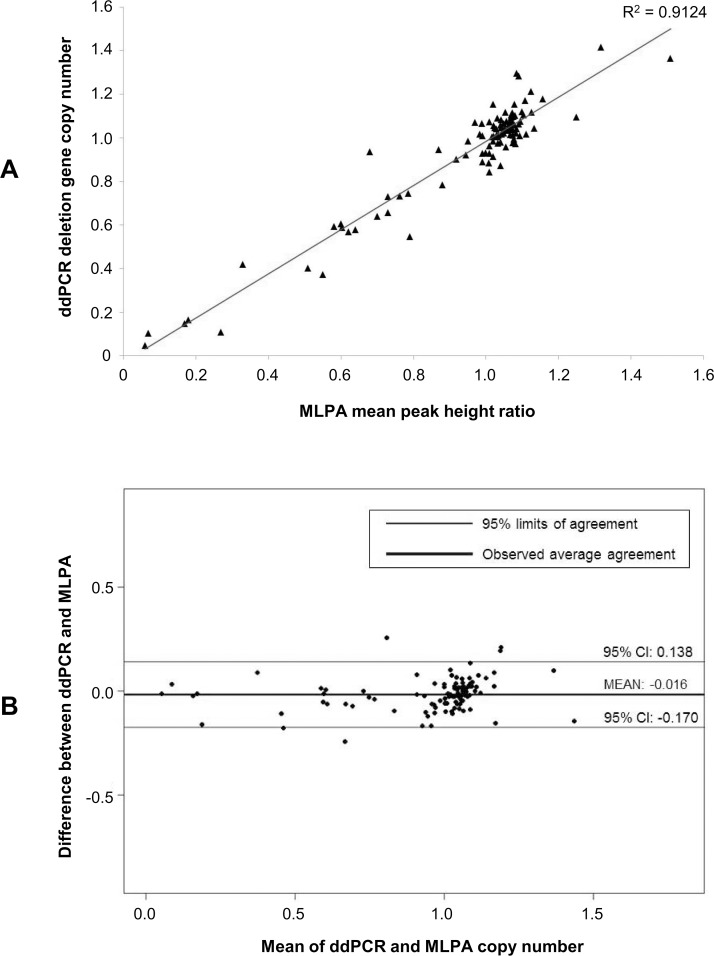
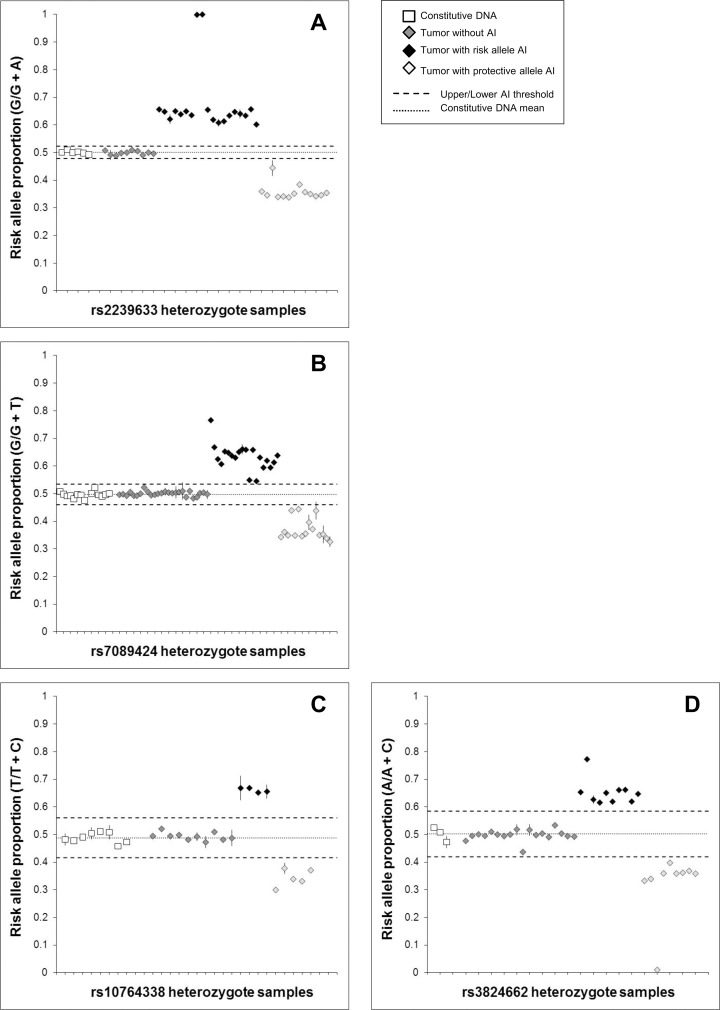
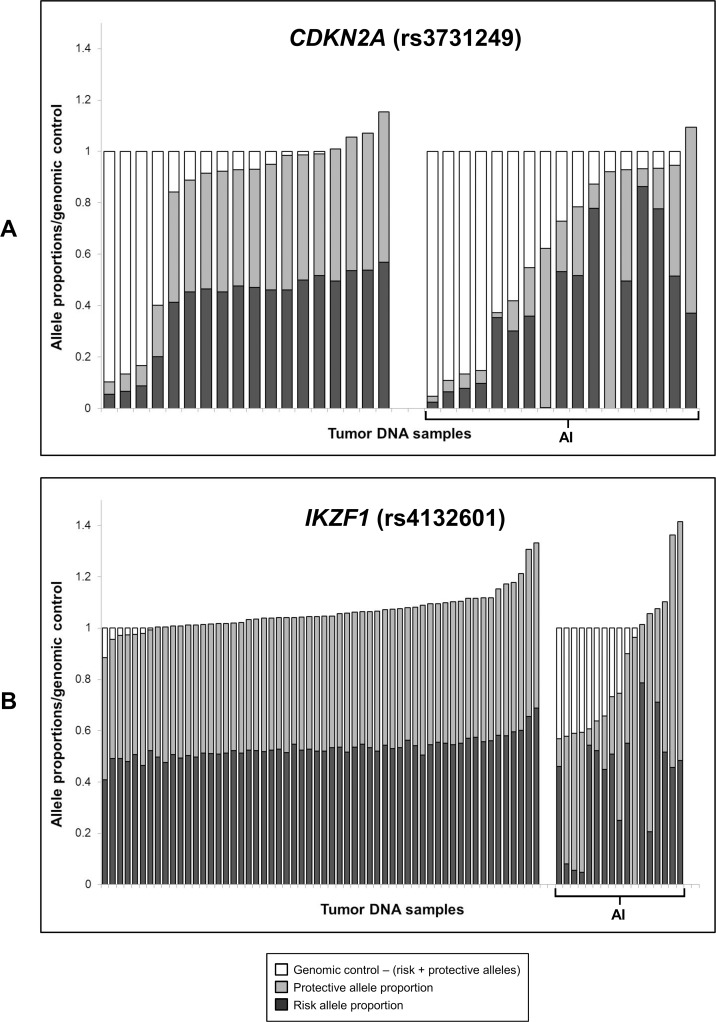
The extent to which heritable genetic variants can affect tumor development has yet to be fully elucidated. Tumor selection of single nucleotide polymorphism (SNP) risk alleles, a phenomenon called preferential allelic imbalance (PAI), has been demonstrated in some cancer types. We developed a novel application of digital PCR termed Somatic Mutation Allelic Ratio Test using Droplet Digital PCR (SMART-ddPCR) for accurate assessment of tumor PAI, and have applied this method to test the hypothesis that heritable SNPs associated with childhood acute lymphoblastic leukemia (ALL) may demonstrate tumor PAI. These SNPs are located at CDKN2A (rs3731217) and IKZF1 (rs4132601), genes frequently lost in ALL, and at CEBPE (rs2239633), ARID5B (rs7089424), PIP4K2A (rs10764338), and GATA3 (rs3824662), genes located on chromosomes gained in high-hyperdiploid ALL. We established thresholds of AI using constitutional DNA from SNP heterozygotes, and subsequently measured allelic copy number in tumor DNA from 19-142 heterozygote samples per SNP locus. We did not find significant tumor PAI at these loci, though CDKN2A and IKZF1 SNPs showed a trend towards preferential selection of the risk allele (p = 0.17 and p = 0.23, respectively). Using a genomic copy number control ddPCR assay, we investigated somatic copy number alterations (SCNA) underlying AI at CDKN2A and IKZF1, revealing a complex range of alterations including homozygous and hemizygous deletions and copy-neutral loss of heterozygosity, with varying degrees of clonality. Copy number estimates from ddPCR showed high agreement with those from multiplex ligation-dependent probe amplification (MLPA) assays. We demonstrate that SMART-ddPCR is a highly accurate method for investigation of tumor PAI and for assessment of the somatic alterations underlying AI. Furthermore, analysis of publicly available data from The Cancer Genome Atlas identified 16 recurrent SCNA loci that contain heritable cancer risk SNPs associated with a matching tumor type, and which represent candidate PAI regions warranting further investigation.
可遗传的基因变异在多大程度上会影响肿瘤的发生发展尚未完全阐明。在某些癌症类型中,已证实存在单核苷酸多态性(SNP)风险等位基因的肿瘤选择现象,即所谓的优先等位基因失衡(PAI)。我们开发了一种新型的数字PCR应用,称为基于液滴数字PCR的体细胞突变等位基因比率测试(SMART-ddPCR),用于准确评估肿瘤PAI,并应用该方法来验证与儿童急性淋巴细胞白血病(ALL)相关的可遗传SNP可能表现出肿瘤PAI这一假设。这些SNP位于CDKN2A(rs3731217)和IKZF1(rs4132601)基因上,这两个基因在ALL中经常缺失;以及CEBPE(rs2239633)、ARID5B(rs7089424)、PIP4K2A(rs10764338)和GATA3(rs3824662)基因上,这些基因位于高超二倍体ALL中获得的染色体上。我们使用SNP杂合子的正常DNA建立了AI阈值,随后测量了每个SNP位点19 - 142个杂合子样本的肿瘤DNA中的等位基因拷贝数。我们在这些位点未发现显著的肿瘤PAI,尽管CDKN2A和IKZF1的SNP显示出优先选择风险等位基因的趋势(分别为p = 0.17和p = 0.23)。使用基因组拷贝数对照ddPCR检测,我们研究了CDKN2A和IKZF1处AI潜在的体细胞拷贝数改变(SCNA),发现了一系列复杂的改变,包括纯合和半合子缺失以及拷贝数中性的杂合性缺失,且克隆性程度各异。ddPCR的拷贝数估计与多重连接依赖探针扩增(MLPA)检测结果高度一致。我们证明SMART-ddPCR是一种用于研究肿瘤PAI和评估AI潜在体细胞改变的高度准确的方法。此外,对癌症基因组图谱公开数据的分析确定了16个反复出现的SCNA位点,这些位点包含与匹配肿瘤类型相关的可遗传癌症风险SNP,代表了值得进一步研究的候选PAI区域。