Li Jia, Poursat Marie-Anne, Drubay Damien, Motz Arnaud, Saci Zohra, Morillon Antonin, Michiels Stefan, Gautheret Daniel
Institute for Integrative Biology of the Cell, CNRS, CEA, Université Paris-Sud, Gif-sur-Yvette, France.
Laboratoire de Mathématique, Université Paris-Sud, Paris, France.
PLoS Comput Biol. 2015 Nov 20;11(11):e1004583. doi: 10.1371/journal.pcbi.1004583. eCollection 2015 Nov.
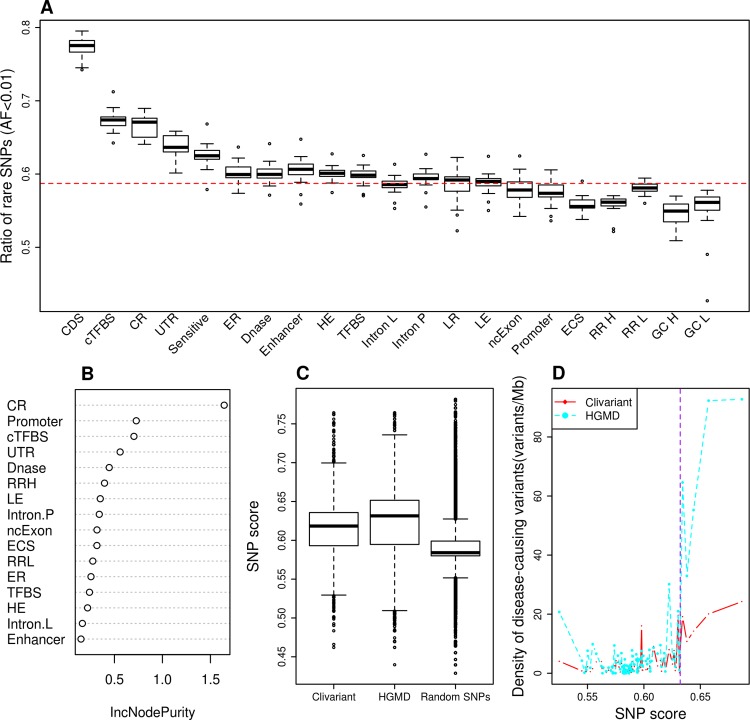
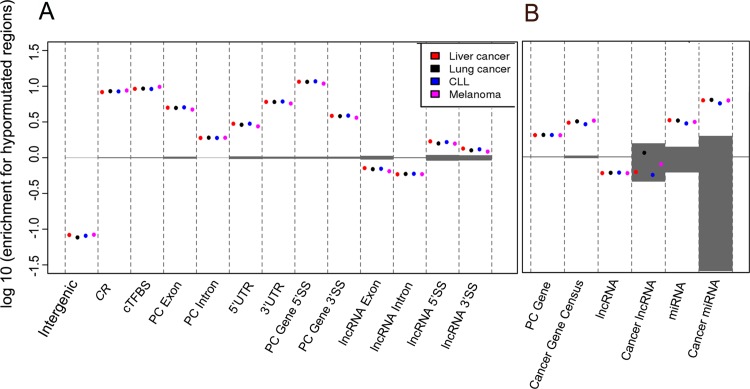
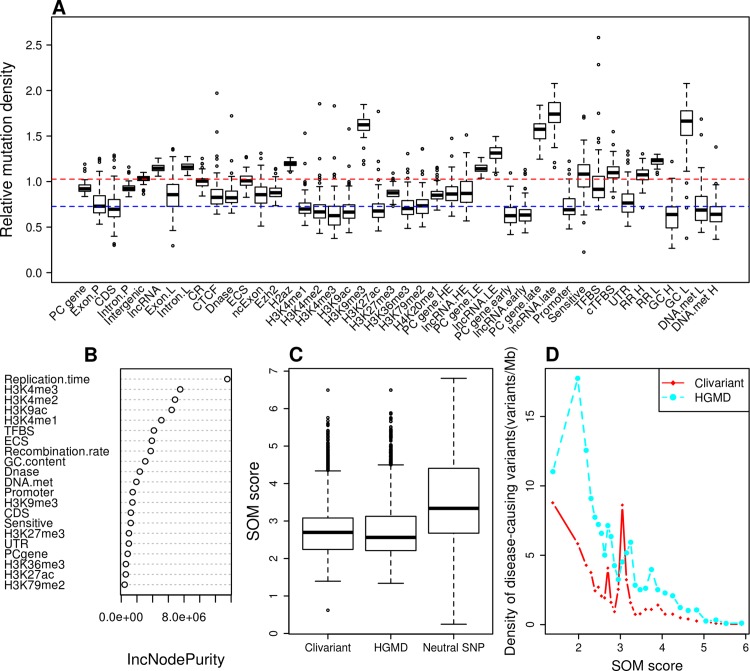
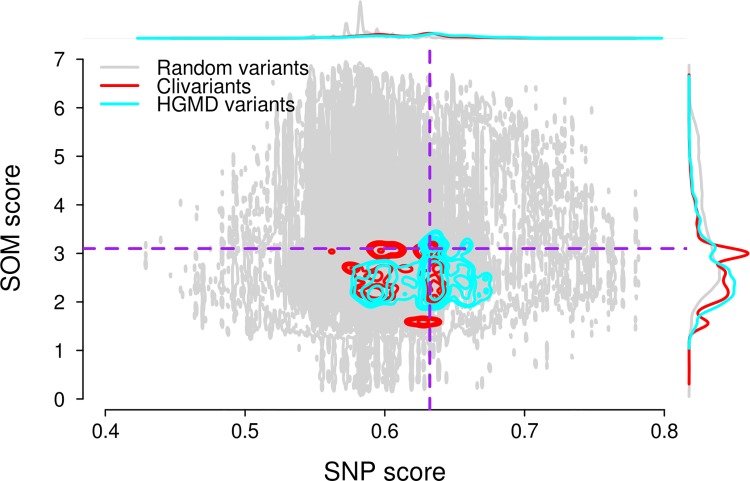
We address here the issue of prioritizing non-coding mutations in the tumoral genome. To this aim, we created two independent computational models. The first (germline) model estimates purifying selection based on population SNP data. The second (somatic) model estimates tumor mutation density based on whole genome tumor sequencing. We show that each model reflects a different set of constraints acting either on the normal or tumor genome, and we identify the specific genome features that most contribute to these constraints. Importantly, we show that the somatic mutation model carries independent functional information that can be used to narrow down the non-coding regions that may be relevant to cancer progression. On this basis, we identify positions in non-coding RNAs and the non-coding parts of mRNAs that are both under purifying selection in the germline and protected from mutation in tumors, thus introducing a new strategy for future detection of cancer driver elements in the expressed non-coding genome.
我们在此探讨肿瘤基因组中非编码突变的优先级问题。为此,我们创建了两个独立的计算模型。第一个(种系)模型基于群体SNP数据估计纯化选择。第二个(体细胞)模型基于全基因组肿瘤测序估计肿瘤突变密度。我们表明,每个模型反映了作用于正常或肿瘤基因组的不同约束集,并且我们确定了对这些约束贡献最大的特定基因组特征。重要的是,我们表明体细胞突变模型携带独立的功能信息,可用于缩小可能与癌症进展相关的非编码区域。在此基础上,我们确定了非编码RNA和mRNA非编码部分中在种系中处于纯化选择且在肿瘤中免受突变影响的位置,从而为未来在表达的非编码基因组中检测癌症驱动元件引入了一种新策略。