Hyvärinen Satu, Jokiranta T Sakari
Department of Bacteriology and Immunology, and Research Programs Unit, Immunobiology, University of Helsinki, Helsinki, Finland.
PLoS One. 2015 Dec 4;10(12):e0143707. doi: 10.1371/journal.pone.0143707. eCollection 2015.
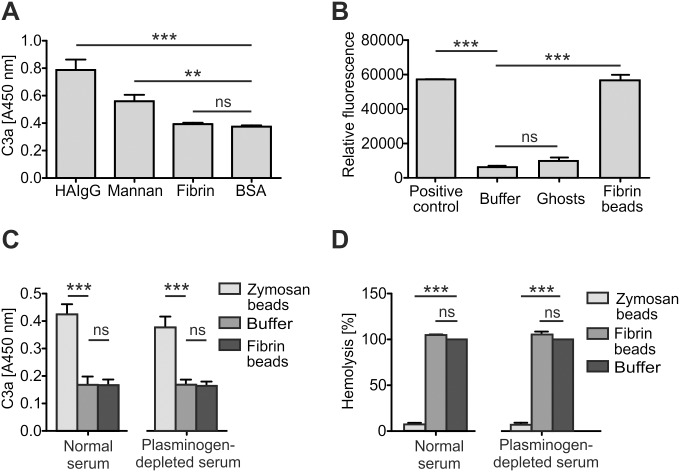
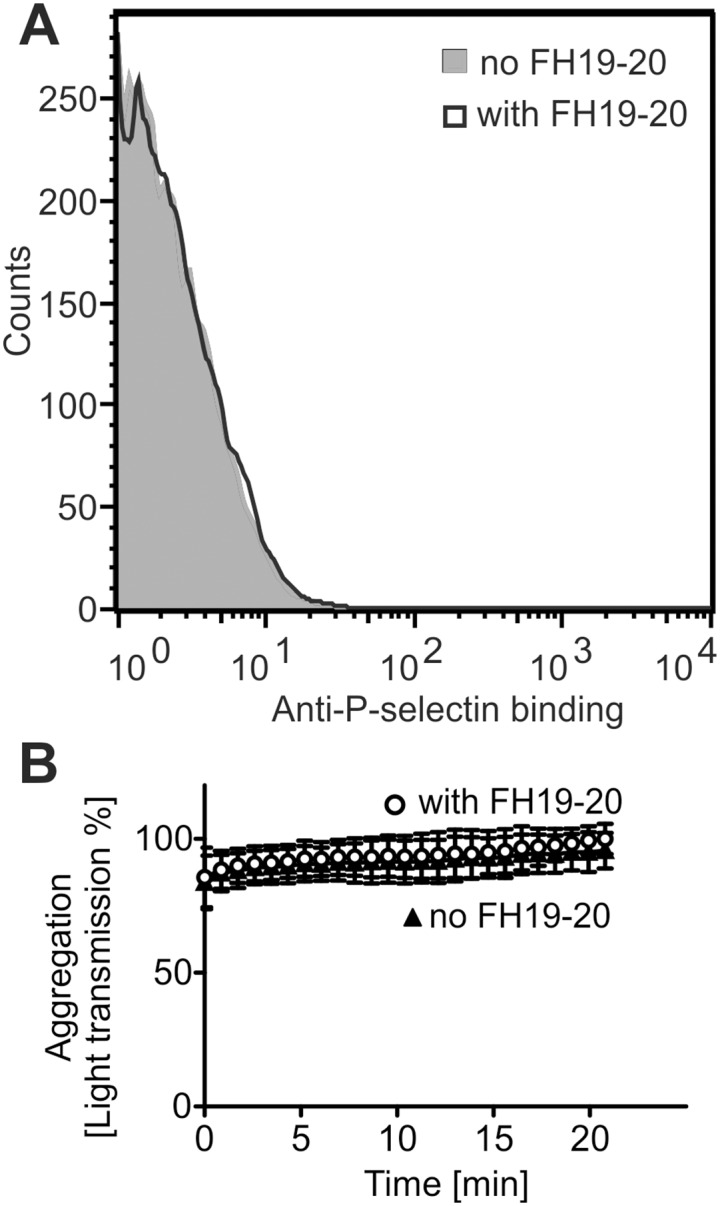
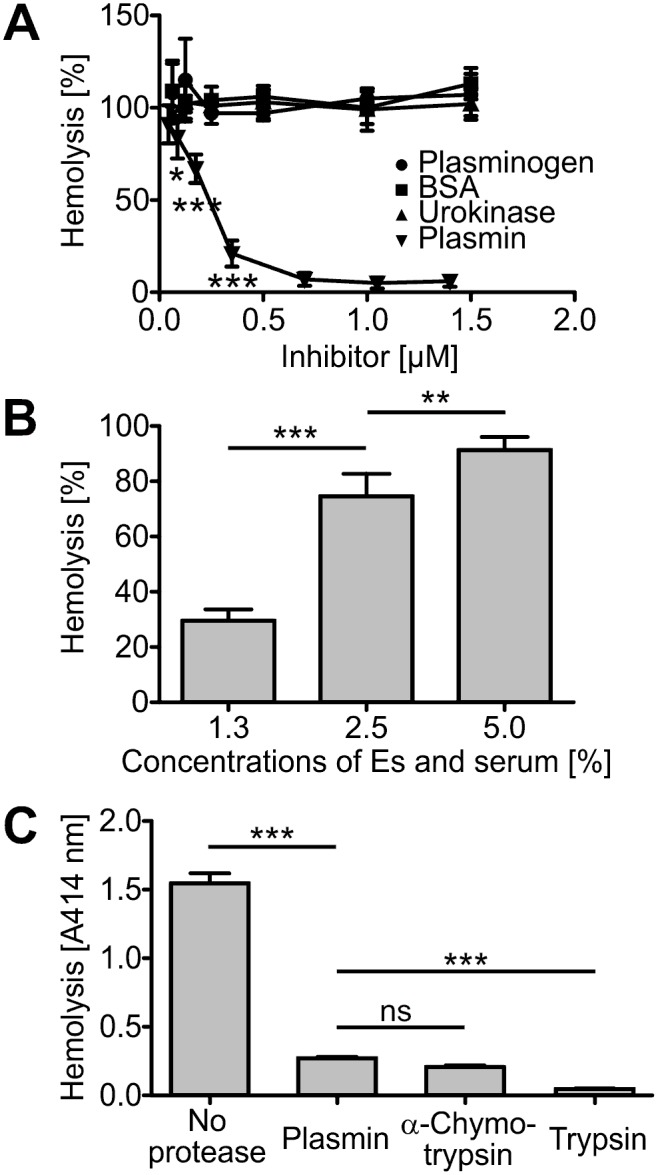
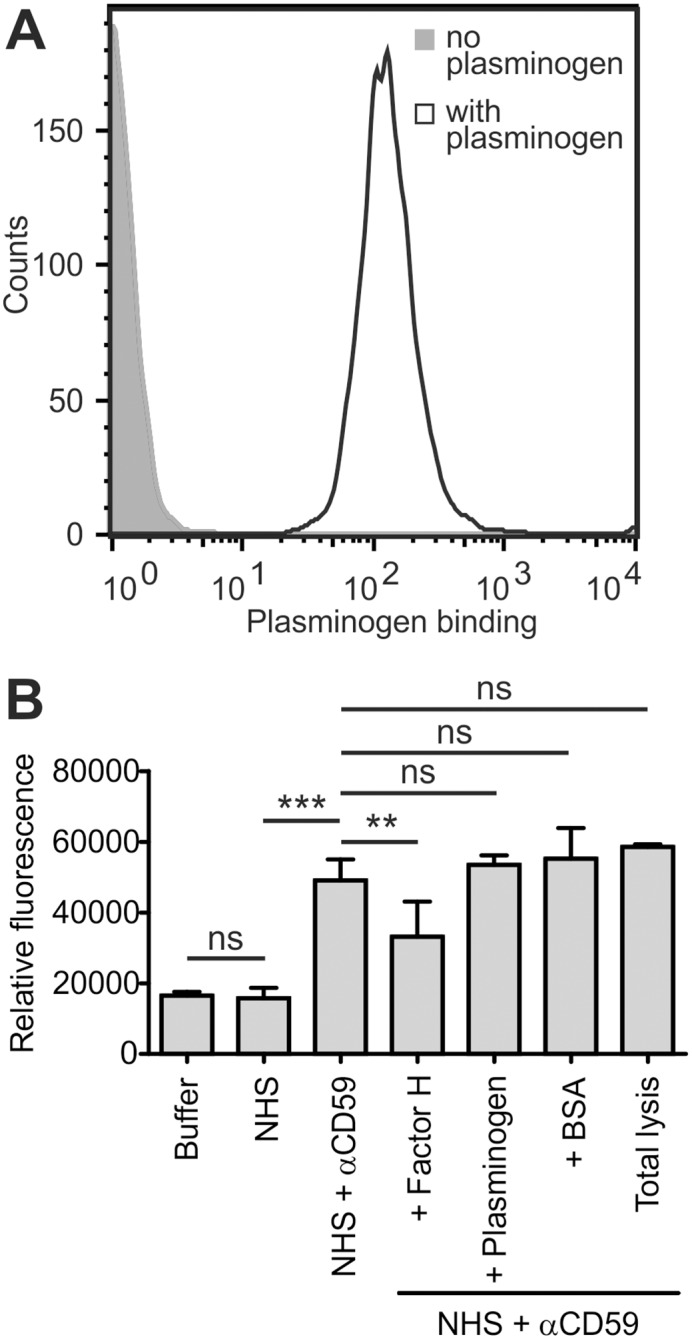
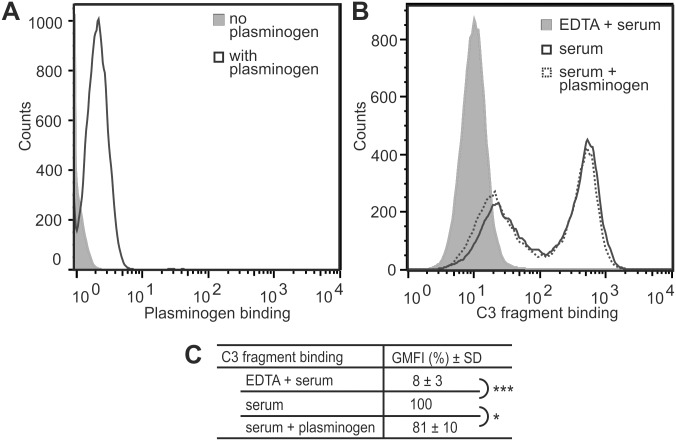
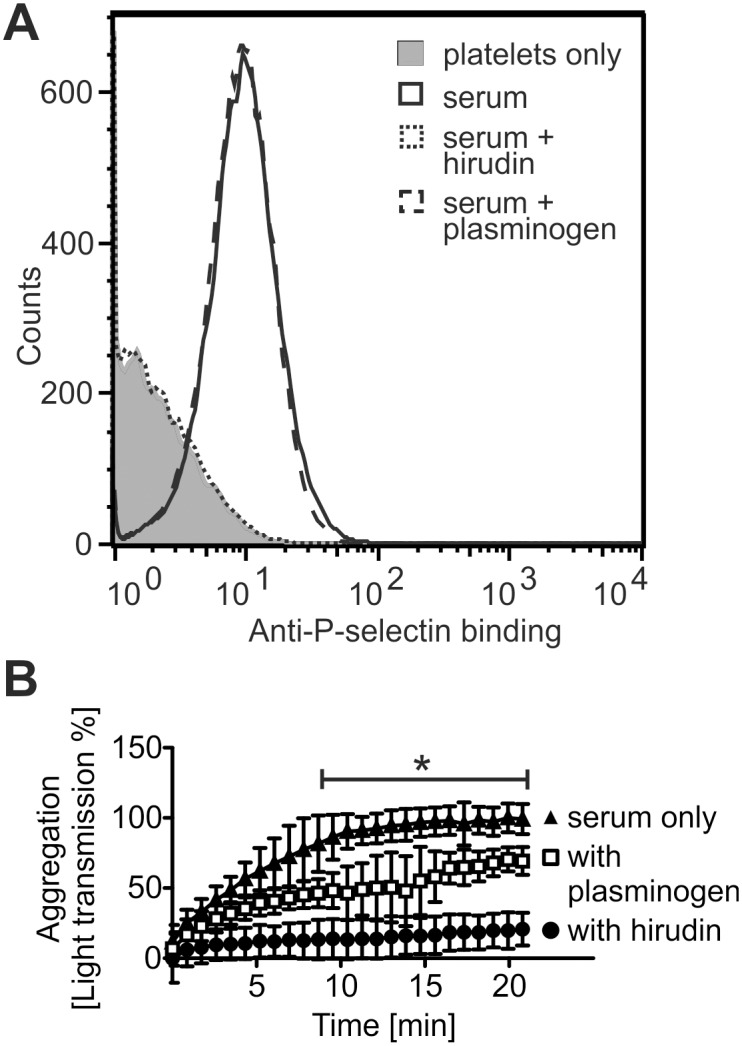
Atypical hemolytic uremic syndrome (aHUS) is a rare, but severe thrombotic microangiopathy. In roughly two thirds of the patients, mutations in complement genes lead to uncontrolled activation of the complement system against self cells. Recently, aHUS patients were described with deficiency of the fibrinolytic protein plasminogen. This zymogen and its protease form plasmin have both been shown to interact with complement proteins in the fluid phase. In this work we studied the potential of plasminogen to restrict complement propagation. In hemolytic assays, plasminogen inhibited complement activation, but only when it had been exogenously activated to plasmin and when it was used at disproportionately high concentrations compared to serum. Addition of only the zymogen plasminogen into serum did not hinder complement-mediated lysis of erythrocytes. Plasminogen could not restrict deposition of complement activation products on endothelial cells either, as was shown with flow cytometry. With platelets, a very weak inhibitory effect on deposition of C3 fragments was observed, but it was considered too weak to be significant for disease pathogenesis. Thus it was concluded that plasminogen is not an important regulator of complement on self cells. Instead, addition of plasminogen was shown to clearly hinder platelet aggregation in serum. This was attributed to plasmin causing disintegration of formed platelet aggregates. We propose that reduced proteolytic activity of plasmin on structures of growing thrombi, rather than on complement activation fragments, explains the association of plasminogen deficiency with aHUS. This adds to the emerging view that factors unrelated to the complement system can also be central to aHUS pathogenesis and suggests that future research on the mechanism of the disease should expand beyond complement dysregulation.
非典型溶血性尿毒症综合征(aHUS)是一种罕见但严重的血栓性微血管病。大约三分之二的患者中,补体基因突变会导致补体系统针对自身细胞的不受控制的激活。最近,有报道称aHUS患者存在纤溶蛋白纤溶酶原缺乏。这种酶原及其蛋白酶形式的纤溶酶均已被证明在液相中与补体蛋白相互作用。在这项研究中,我们研究了纤溶酶原限制补体传播的潜力。在溶血试验中,纤溶酶原抑制补体激活,但仅在其被外源性激活为纤溶酶且与血清相比使用的浓度过高时才会如此。仅向血清中添加酶原纤溶酶原并不会阻碍补体介导的红细胞裂解。流式细胞术显示,纤溶酶原也无法限制补体激活产物在内皮细胞上的沉积。对于血小板,观察到对C3片段沉积有非常微弱的抑制作用,但认为其过于微弱,对疾病发病机制无显著意义。因此得出结论,纤溶酶原不是自身细胞上补体的重要调节因子。相反,研究表明添加纤溶酶原会明显阻碍血清中的血小板聚集。这归因于纤溶酶导致已形成的血小板聚集体解体。我们提出,纤溶酶对正在生长的血栓结构而非补体激活片段的蛋白水解活性降低,解释了纤溶酶原缺乏与aHUS的关联。这进一步支持了一种新出现的观点,即与补体系统无关的因素也可能是aHUS发病机制的核心,并表明未来对该疾病机制的研究应超越补体失调的范畴。