Wolfien Markus, Rimmbach Christian, Schmitz Ulf, Jung Julia Jeannine, Krebs Stefan, Steinhoff Gustav, David Robert, Wolkenhauer Olaf
Department of Systems Biology and Bioinformatics, University of Rostock, 18057, Rostock, Germany.
Reference und Translation Center for Cardiac Stem Cell Therapy (RTC), University of Rostock, Rostock, 18057, Germany.
BMC Bioinformatics. 2016 Jan 6;17:21. doi: 10.1186/s12859-015-0873-9.
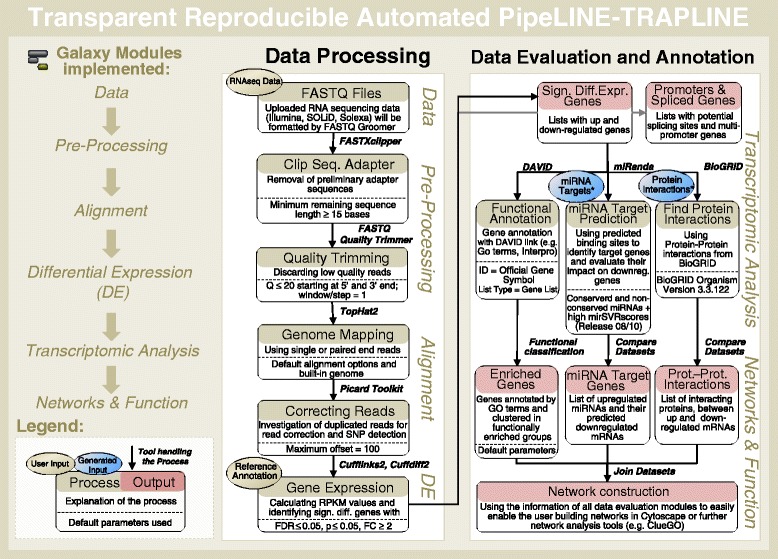
Technical advances in Next Generation Sequencing (NGS) provide a means to acquire deeper insights into cellular functions. The lack of standardized and automated methodologies poses a challenge for the analysis and interpretation of RNA sequencing data. We critically compare and evaluate state-of-the-art bioinformatics approaches and present a workflow that integrates the best performing data analysis, data evaluation and annotation methods in a Transparent, Reproducible and Automated PipeLINE (TRAPLINE) for RNA sequencing data processing (suitable for Illumina, SOLiD and Solexa).
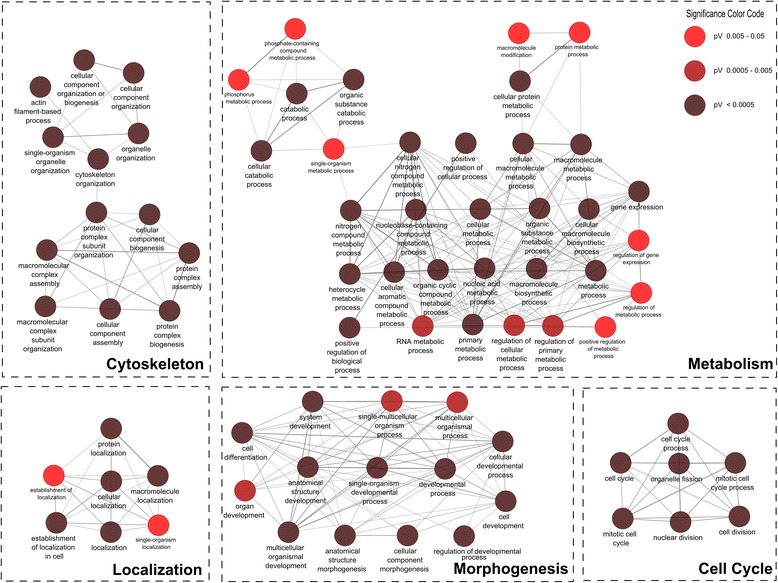
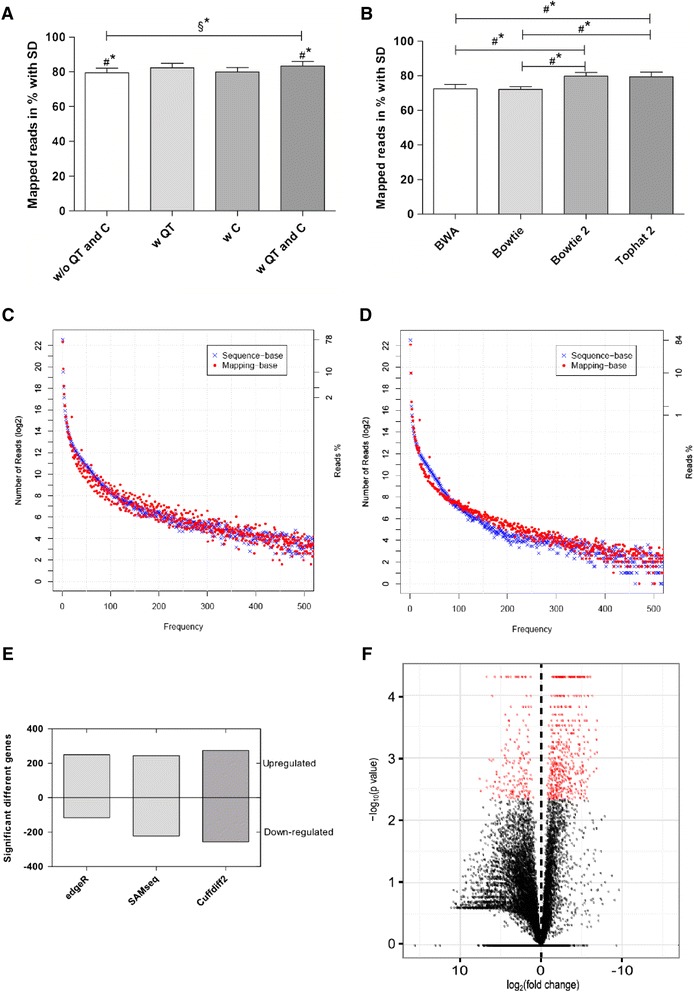
Comparative transcriptomics analyses with TRAPLINE result in a set of differentially expressed genes, their corresponding protein-protein interactions, splice variants, promoter activity, predicted miRNA-target interactions and files for single nucleotide polymorphism (SNP) calling. The obtained results are combined into a single file for downstream analysis such as network construction. We demonstrate the value of the proposed pipeline by characterizing the transcriptome of our recently described stem cell derived antibiotic selected cardiac bodies ('aCaBs').
TRAPLINE supports NGS-based research by providing a workflow that requires no bioinformatics skills, decreases the processing time of the analysis and works in the cloud. The pipeline is implemented in the biomedical research platform Galaxy and is freely accessible via www.sbi.uni-rostock.de/RNAseqTRAPLINE or the specific Galaxy manual page (https://usegalaxy.org/u/mwolfien/p/trapline---manual).
新一代测序(NGS)技术的进步为深入了解细胞功能提供了一种手段。缺乏标准化和自动化的方法给RNA测序数据的分析和解释带来了挑战。我们严格比较和评估了当前最先进的生物信息学方法,并提出了一种工作流程,该流程将性能最佳的数据分析、数据评估和注释方法集成到一个用于RNA测序数据处理的透明、可重复和自动化管道(TRAPLINE)中(适用于Illumina、SOLiD和Solexa)。
使用TRAPLINE进行的比较转录组学分析产生了一组差异表达基因、它们相应的蛋白质-蛋白质相互作用、剪接变体、启动子活性、预测的miRNA-靶标相互作用以及用于单核苷酸多态性(SNP)检测的文件。所获得的结果被合并到一个单一文件中,用于下游分析,如网络构建。我们通过表征我们最近描述的干细胞衍生的抗生素筛选心肌小体(“aCaBs”)的转录组来证明所提出管道的价值。
TRAPLINE通过提供一种无需生物信息学技能、减少分析处理时间且可在云端运行的工作流程来支持基于NGS的研究。该管道在生物医学研究平台Galaxy中实现,可通过www.sbi.uni-rostock.de/RNAseqTRAPLINE或特定的Galaxy手册页面(https://usegalaxy.org/u/mwolfien/p/trapline---manual)免费访问。