Meher Prabina Kumar, Sahu Tanmaya Kumar, Rao Atmakuri Ramakrishna
Division of Statistical Genetics, Indian Agricultural Statistics Research Institute, New Delhi, 110 012 India.
Centre for Agricultural Bioinformatics, Indian Agricultural Statistics Research Institute, New Delhi, 110 012 India.
BioData Min. 2016 Jan 22;9:4. doi: 10.1186/s13040-016-0086-4. eCollection 2016.
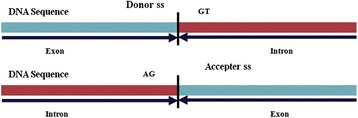
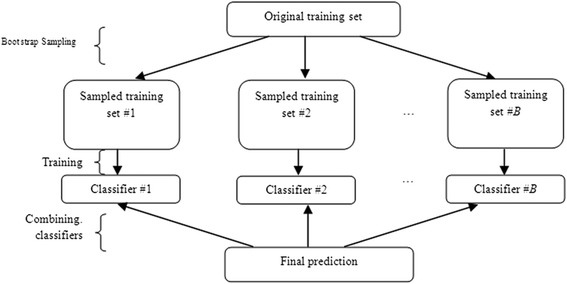
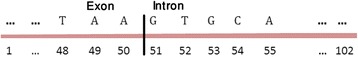
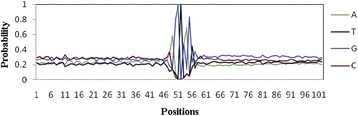
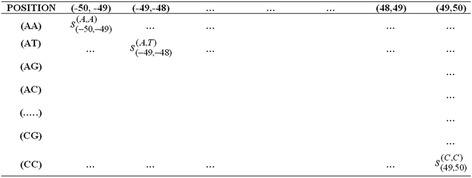
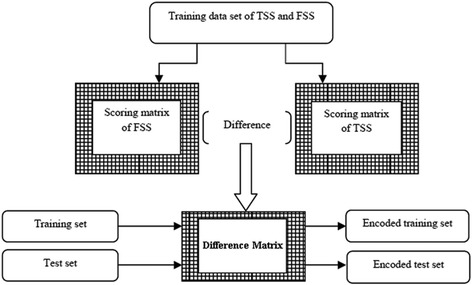
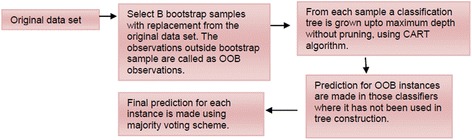
Detection of splice sites plays a key role for predicting the gene structure and thus development of efficient analytical methods for splice site prediction is vital. This paper presents a novel sequence encoding approach based on the adjacent di-nucleotide dependencies in which the donor splice site motifs are encoded into numeric vectors. The encoded vectors are then used as input in Random Forest (RF), Support Vector Machines (SVM) and Artificial Neural Network (ANN), Bagging, Boosting, Logistic regression, kNN and Naïve Bayes classifiers for prediction of donor splice sites.
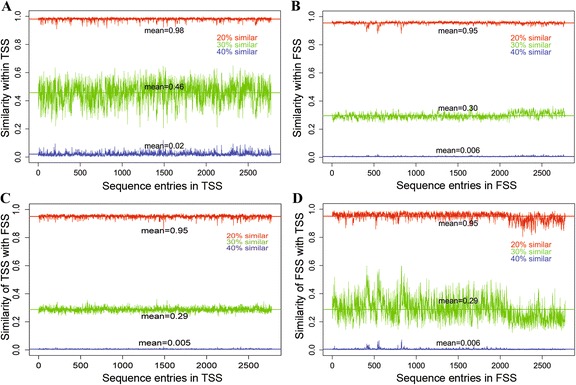
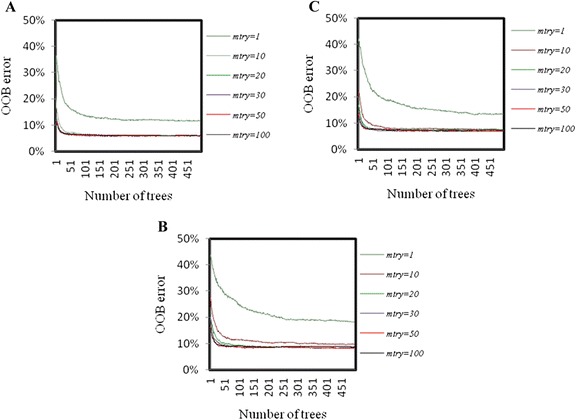
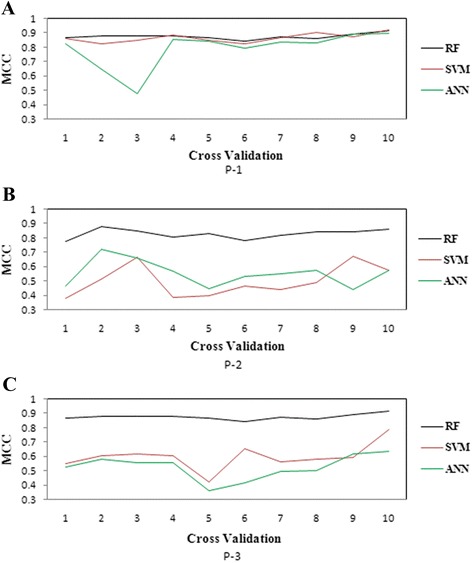
The performance of the proposed approach is evaluated on the donor splice site sequence data of Homo sapiens, collected from Homo Sapiens Splice Sites Dataset (HS3D). The results showed that RF outperformed all the considered classifiers. Besides, RF achieved higher prediction accuracy than the existing methods viz., MEM, MDD, WMM, MM1, NNSplice and SpliceView, while compared using an independent test dataset.
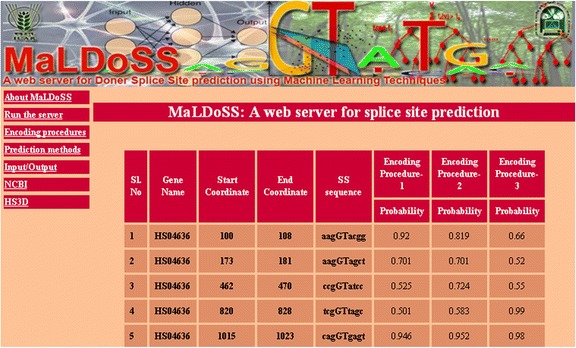
Based on the proposed approach, we have developed an online prediction server (MaLDoSS) to help the biological community in predicting the donor splice sites. The server is made freely available at http://cabgrid.res.in:8080/maldoss. Due to computational feasibility and high prediction accuracy, the proposed approach is believed to help in predicting the eukaryotic gene structure.
剪接位点的检测对于预测基因结构起着关键作用,因此开发高效的剪接位点预测分析方法至关重要。本文提出了一种基于相邻二核苷酸依赖性的新型序列编码方法,其中供体剪接位点基序被编码为数字向量。然后将编码后的向量用作随机森林(RF)、支持向量机(SVM)、人工神经网络(ANN)、装袋法、提升法、逻辑回归、k近邻和朴素贝叶斯分类器的输入,用于预测供体剪接位点。
在所提出的方法的性能在从智人剪接位点数据集(HS3D)收集的智人的供体剪接位点序列数据上进行了评估。结果表明,随机森林在所有考虑的分类器中表现最佳。此外,在使用独立测试数据集进行比较时,随机森林比现有方法即MEM、MDD、WMM、MM1、NNSplice和SpliceView实现了更高的预测准确率。
基于所提出的方法,我们开发了一个在线预测服务器(MaLDoSS),以帮助生物界预测供体剪接位点。该服务器可在http://cabgrid.res.in:8080/maldoss免费获取。由于计算可行性和高预测准确率,所提出的方法被认为有助于预测真核基因结构。