Wang Cheng, Dong Da, Wang Haoshu, Müller Karin, Qin Yong, Wang Hailong, Wu Weixiang
Zhejiang Province Key Laboratory for Water Pollution Control and Environmental Safety Technology, Institute of Environmental Science and Technology, Zhejiang University, 866 Yuhangtang Road, Hangzhou, 310058 China.
Zhejiang Province Key Laboratory for Water Pollution Control and Environmental Safety Technology, Institute of Environmental Science and Technology, Zhejiang University, 866 Yuhangtang Road, Hangzhou, 310058 China ; Key Laboratory of Soil Contamination Bioremediation of Zhejiang Province, School of Environmental and Resource Sciences, Zhejiang A & F University, Lin'an, Hangzhou, 311300 China.
Biotechnol Biofuels. 2016 Jan 29;9:22. doi: 10.1186/s13068-016-0440-2. eCollection 2016.
Compost habitats sustain a vast ensemble of microbes specializing in the degradation of lignocellulosic plant materials and are thus important both for their roles in the global carbon cycle and as potential sources of biochemical catalysts for advanced biofuels production. Studies have revealed substantial diversity in compost microbiomes, yet how this diversity relates to functions and even to the genes encoding lignocellulolytic enzymes remains obscure. Here, we used a metagenomic analysis of the rice straw-adapted (RSA) microbial consortia enriched from compost ecosystems to decipher the systematic and functional contexts within such a distinctive microbiome.
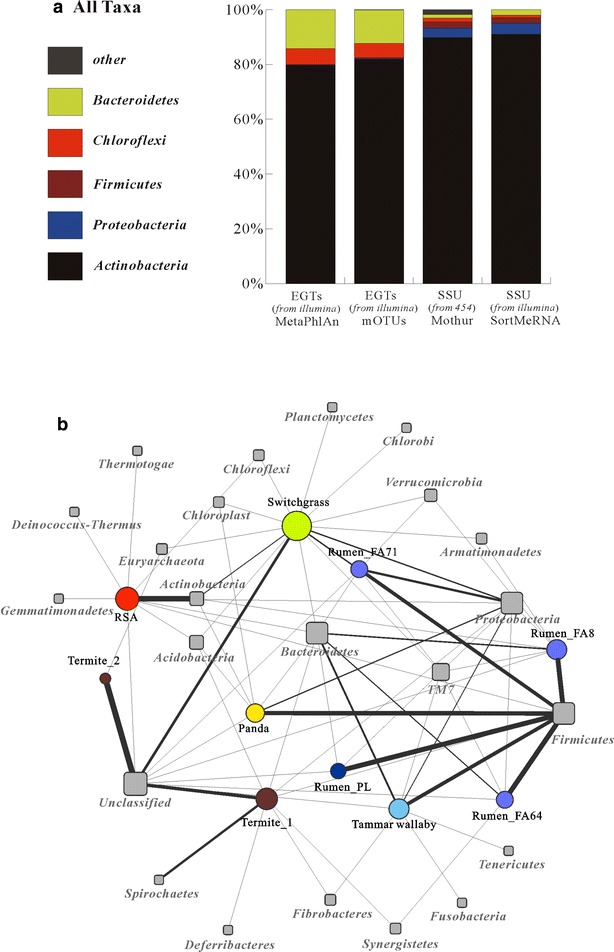
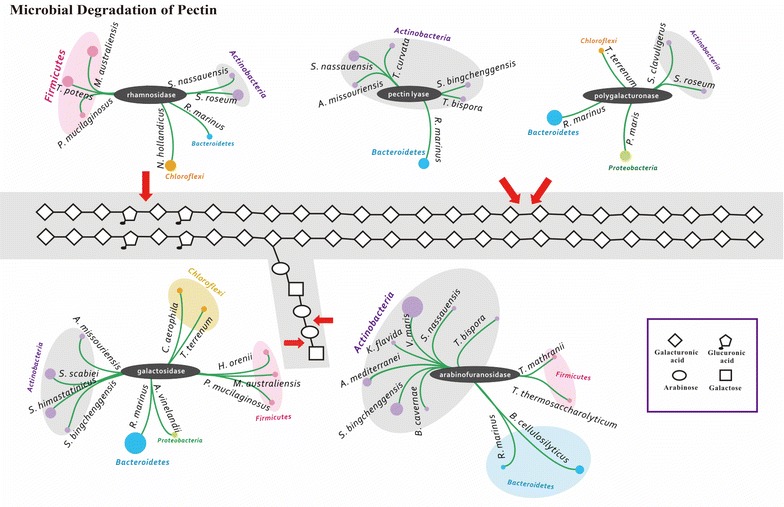
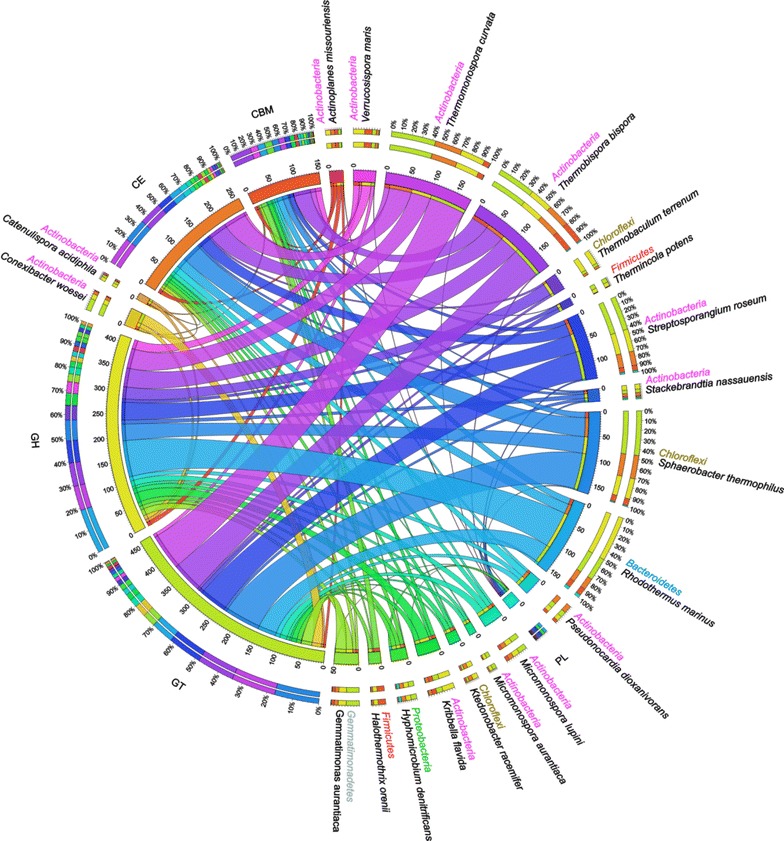
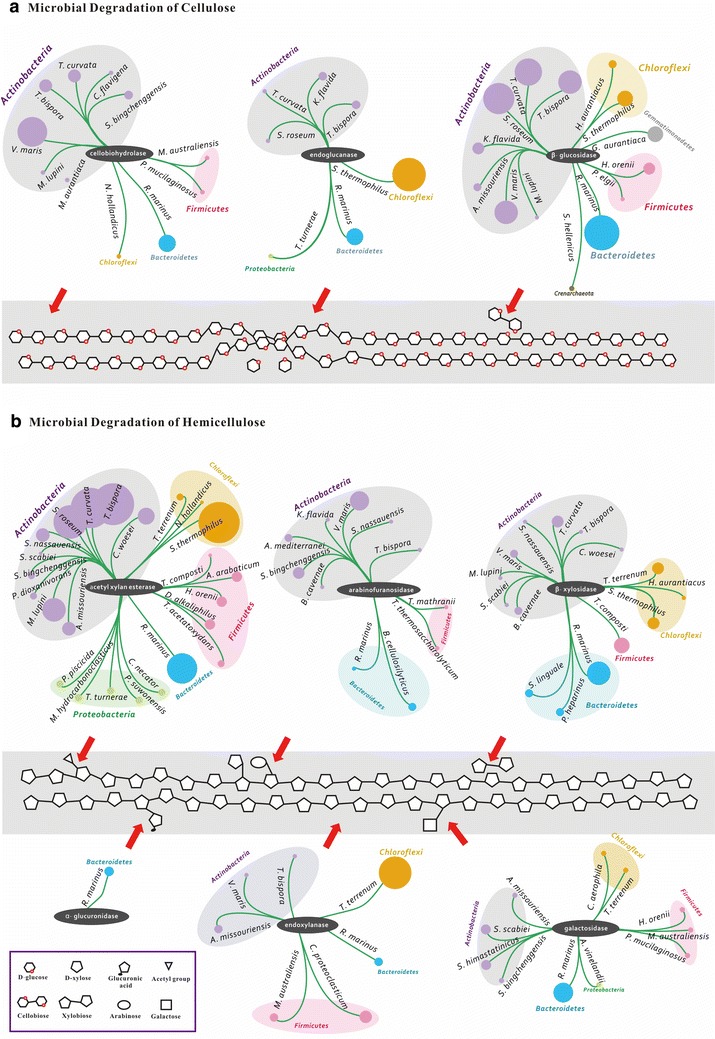
Analyses of the 16S pyrotag library and 5 Gbp of metagenomic sequence showed that the phylum Actinobacteria was the predominant group among the Bacteria in the RSA consortia, followed by Proteobacteria, Firmicutes, Chloroflexi, and Bacteroidetes. The CAZymes profile revealed that CAZyme genes in the RSA consortia were also widely distributed within these bacterial phyla. Strikingly, about 46.1 % of CAZyme genes were from actinomycetal communities, which harbored a substantially expanded catalog of the cellobiohydrolase, β-glucosidase, acetyl xylan esterase, arabinofuranosidase, pectin lyase, and ligninase genes. Among these communities, a variety of previously unrecognized species was found, which reveals a greater ecological functional diversity of thermophilic Actinobacteria than previously assumed.
These data underline the pivotal role of thermophilic Actinobacteria in lignocellulose biodegradation processes in the compost habitat. Besides revealing a new benchmark for microbial enzymatic deconstruction of lignocelluloses, the results suggest that actinomycetes found in compost ecosystems are potential candidates for mining efficient lignocellulosic enzymes in the biofuel industry.
堆肥生境中存在大量专门降解木质纤维素植物材料的微生物群落,因此它们在全球碳循环中发挥着重要作用,并且作为先进生物燃料生产中生化催化剂的潜在来源也很重要。研究揭示了堆肥微生物群落具有丰富的多样性,但这种多样性与功能以及与编码木质纤维素酶的基因之间的关系仍不清楚。在这里,我们对从堆肥生态系统中富集的适应稻草(RSA)微生物群落进行了宏基因组分析,以解读这种独特微生物群落中的系统和功能背景。
对16S焦磷酸测序文库和5 Gbp宏基因组序列的分析表明,放线菌门是RSA群落中细菌的主要类群,其次是变形菌门、厚壁菌门、绿弯菌门和拟杆菌门。碳水化合物活性酶(CAZyme)谱显示,RSA群落中的CAZyme基因也广泛分布于这些细菌门中。引人注目的是,约46.1%的CAZyme基因来自放线菌群落,这些群落拥有大量扩展的纤维二糖水解酶、β-葡萄糖苷酶、乙酰木聚糖酯酶、阿拉伯呋喃糖苷酶、果胶裂解酶和木质素酶基因目录。在这些群落中,发现了多种以前未被识别的物种,这揭示了嗜热放线菌的生态功能多样性比以前认为的更大。
这些数据强调了嗜热放线菌在堆肥生境中木质纤维素生物降解过程中的关键作用。除了揭示木质纤维素微生物酶解的新基准外,结果还表明,在堆肥生态系统中发现的放线菌是生物燃料行业中挖掘高效木质纤维素酶的潜在候选者。