Ji Jiadong, Yuan Zhongshang, Zhang Xiaoshuai, Xue Fuzhong
Department of Biostatistics, School of Public Health, Shandong University, PO Box 100, Jinan, 250012, Shandong, China.
BMC Bioinformatics. 2016 Feb 12;17:86. doi: 10.1186/s12859-016-0916-x.
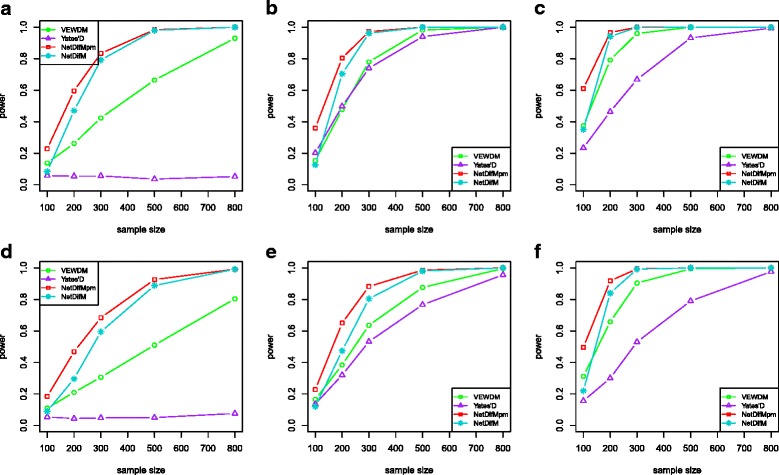
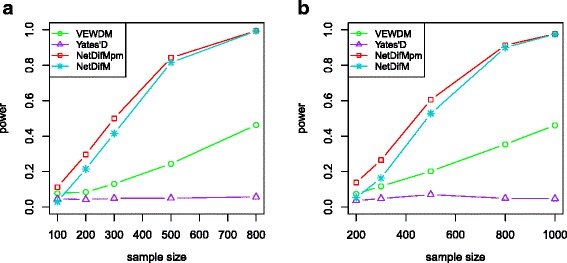
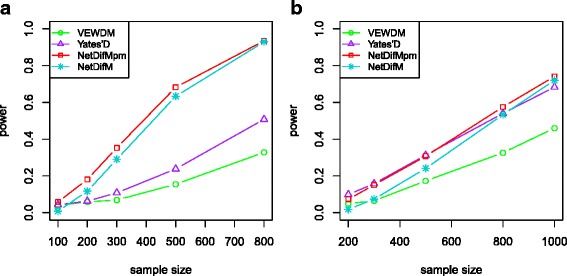
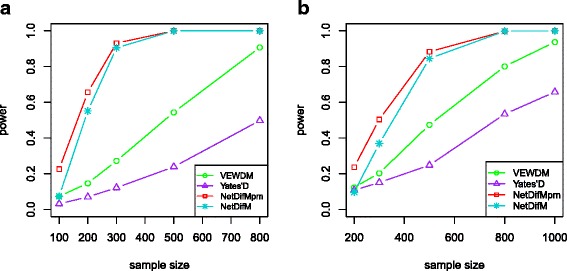
Complex disease is largely determined by a number of biomolecules interwoven into networks, rather than a single biomolecule. A key but inadequately addressed issue is how to test possible differences of the networks between two groups. Group-level comparison of network properties may shed light on underlying disease mechanisms and benefit the design of drug targets for complex diseases. We therefore proposed a powerful score-based statistic to detect group difference in weighted networks, which simultaneously capture the vertex changes and edge changes.
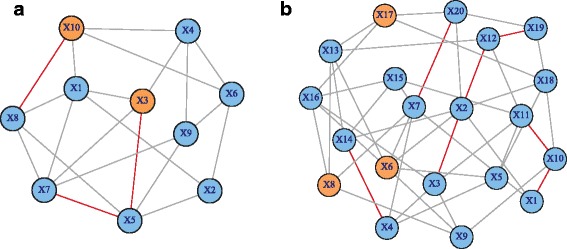
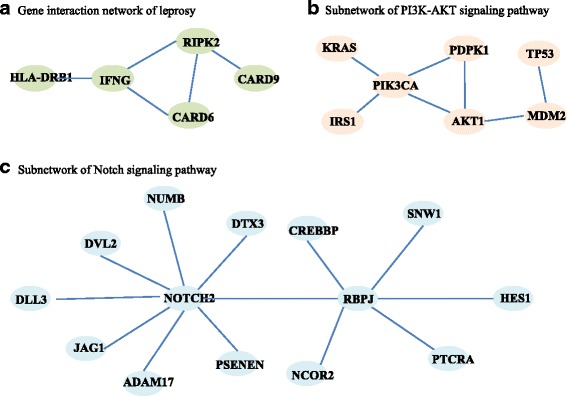
Simulation studies indicated that the proposed network difference measure (NetDifM) was stable and outperformed other methods existed, under various sample sizes and network topology structure. One application to real data about GWAS of leprosy successfully identified the specific gene interaction network contributing to leprosy. For additional gene expression data of ovarian cancer, two candidate subnetworks, PI3K-AKT and Notch signaling pathways, were considered and identified respectively.
The proposed method, accounting for the vertex changes and edge changes simultaneously, is valid and powerful to capture the group difference of biological networks.
复杂疾病很大程度上由交织在网络中的多种生物分子决定,而非单个生物分子。一个关键但尚未得到充分解决的问题是如何测试两组之间网络的可能差异。网络属性的组间比较可能有助于揭示潜在的疾病机制,并有利于复杂疾病药物靶点的设计。因此,我们提出了一种强大的基于分数的统计量来检测加权网络中的组间差异,该统计量同时捕捉顶点变化和边变化。
模拟研究表明,在各种样本量和网络拓扑结构下,所提出的网络差异度量(NetDifM)是稳定的,并且优于其他现有方法。对麻风病全基因组关联研究的真实数据进行的一项应用成功识别出了导致麻风病的特定基因相互作用网络。对于卵巢癌的额外基因表达数据,分别考虑并识别了两个候选子网,即PI3K - AKT和Notch信号通路。
所提出的方法同时考虑顶点变化和边变化,在捕捉生物网络的组间差异方面是有效且强大的。