Zou Chenchen, Zhang Yuping, Ouyang Zhengqing
The Jackson Laboratory for Genomic Medicine, Farmington, 06032, CT, USA.
Department of Statistics, University of Connecticut, Storrs, 06269, CT, USA.
Genome Biol. 2016 Mar 2;17:40. doi: 10.1186/s13059-016-0896-1.
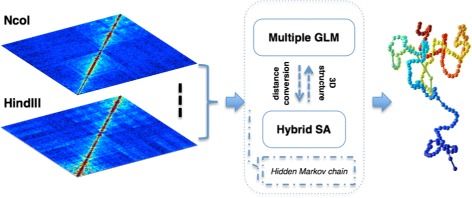
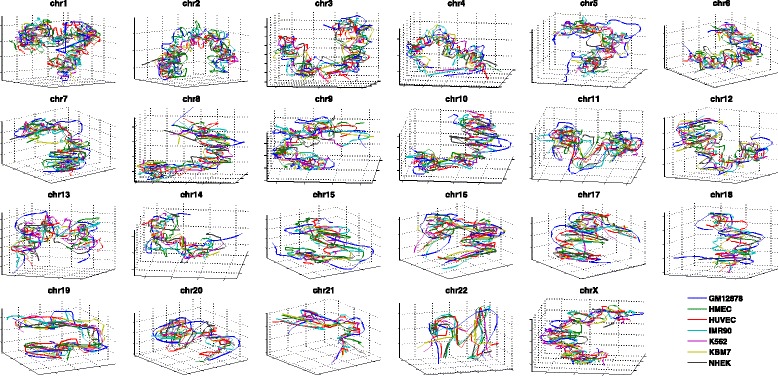
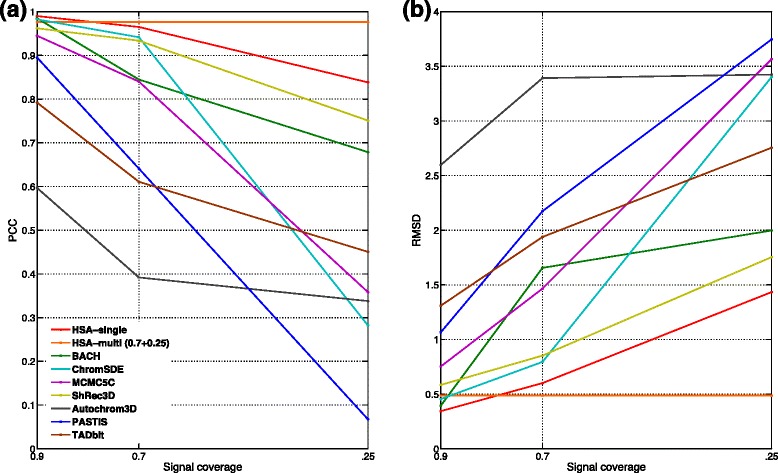
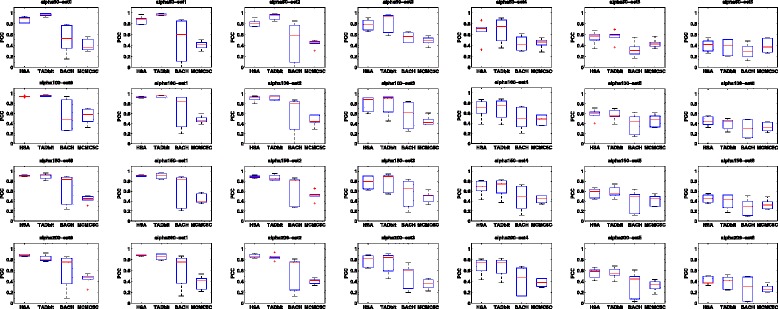
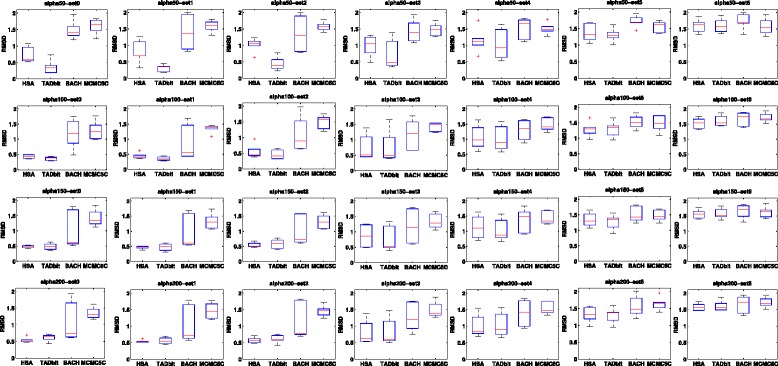
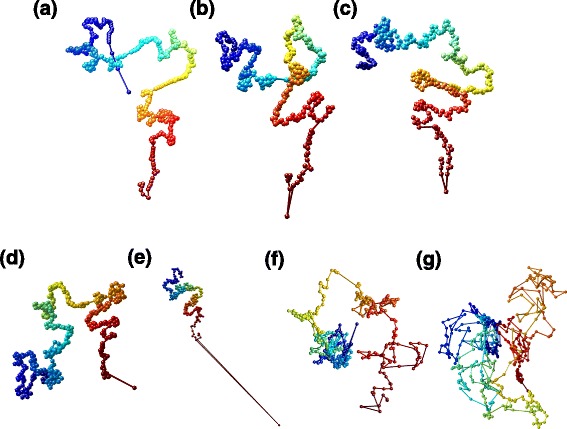
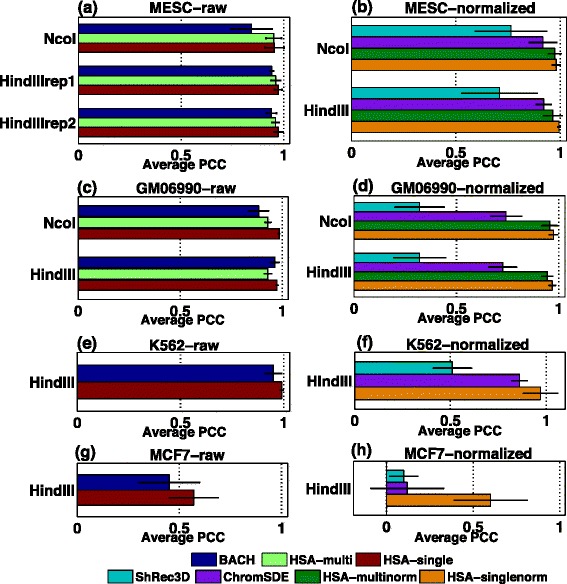
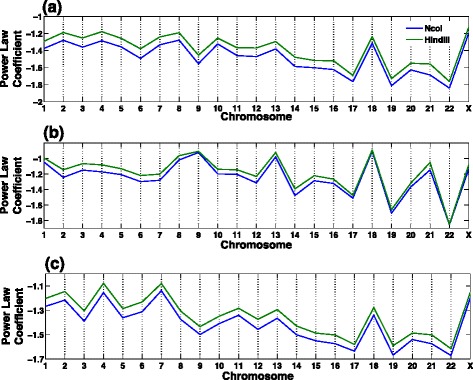
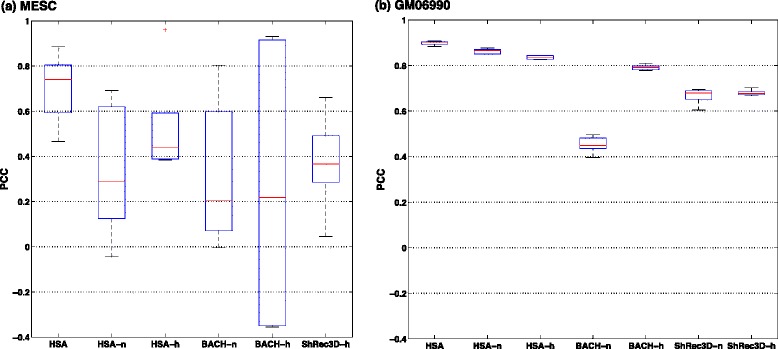
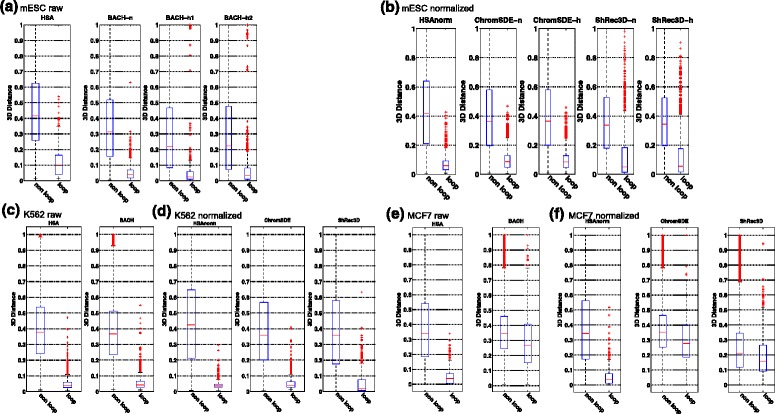
Genome-wide 3C technologies (Hi-C) are being increasingly employed to study three-dimensional (3D) genome conformations. Existing computational approaches are unable to integrate accumulating data to facilitate studying 3D chromatin structure and function. We present HSA ( http://ouyanglab.jax.org/hsa/ ), a flexible tool that jointly analyzes multiple contact maps to infer 3D chromatin structure at the genome scale. HSA globally searches the latent structure underlying different cleavage footprints. Its robustness and accuracy outperform or rival existing tools on extensive simulations and orthogonal experiment validations. Applying HSA to recent in situ Hi-C data, we found the 3D chromatin structures are highly conserved across various human cell types.
全基因组3C技术(Hi-C)正越来越多地用于研究三维(3D)基因组构象。现有的计算方法无法整合不断积累的数据以促进对3D染色质结构和功能的研究。我们展示了HSA(http://ouyanglab.jax.org/hsa/),这是一个灵活的工具,可联合分析多个接触图谱以推断基因组规模的3D染色质结构。HSA全局搜索不同切割足迹背后的潜在结构。在广泛的模拟和正交实验验证中,其稳健性和准确性优于或可与现有工具相媲美。将HSA应用于最近的原位Hi-C数据,我们发现3D染色质结构在各种人类细胞类型中高度保守。