Cai Binghuang, Jiang Xia
Department of Biomedical Informatics, School of Medicine, University of Pittsburgh, Pittsburgh, PA, 15206-3701, USA.
BMC Bioinformatics. 2016 Mar 3;17:116. doi: 10.1186/s12859-016-0959-z.
Ubiquitination is a very important process in protein post-translational modification, which has been widely investigated by biology scientists and researchers. Different experimental and computational methods have been developed to identify the ubiquitination sites in protein sequences. This paper aims at exploring computational machine learning methods for the prediction of ubiquitination sites using the physicochemical properties (PCPs) of amino acids in the protein sequences.
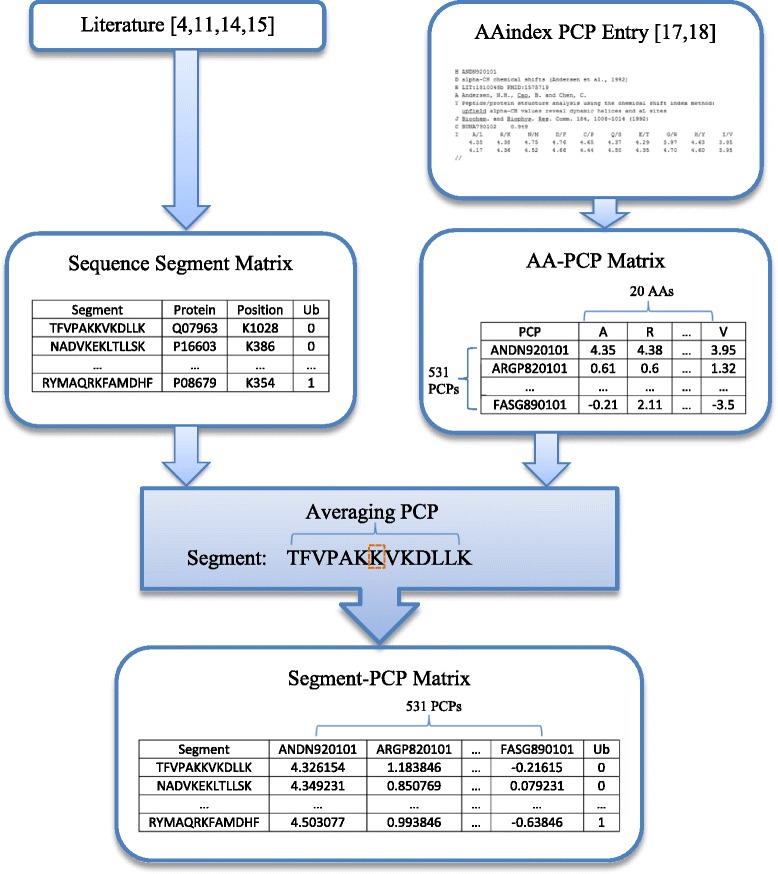
We first establish six different ubiquitination data sets, whose records contain both ubiquitination sites and non-ubiquitination sites in variant numbers of protein sequence segments. In particular, to establish such data sets, protein sequence segments are extracted from the original protein sequences used in four published papers on ubiquitination, while 531 PCP features of each extracted protein sequence segment are calculated based on PCP values from AAindex (Amino Acid index database) by averaging PCP values of all amino acids on each segment. Various computational machine-learning methods, including four Bayesian network methods (i.e., Naïve Bayes (NB), Feature Selection NB (FSNB), Model Averaged NB (MANB), and Efficient Bayesian Multivariate Classifier (EBMC)) and three regression methods (i.e., Support Vector Machine (SVM), Logistic Regression (LR), and Least Absolute Shrinkage and Selection Operator (LASSO)), are then applied to the six established segment-PCP data sets. Five-fold cross-validation and the Area Under Receiver Operating Characteristic Curve (AUROC) are employed to evaluate the ubiquitination prediction performance of each method. Results demonstrate that the PCP data of protein sequences contain information that could be mined by machine learning methods for ubiquitination site prediction. The comparative results show that EBMC, SVM and LR perform better than other methods, and EBMC is the only method that can get AUCs greater than or equal to 0.6 for the six established data sets. Results also show EBMC tends to perform better for larger data.
Machine learning methods have been employed for the ubiquitination site prediction based on physicochemical properties of amino acids on protein sequences. Results demonstrate the effectiveness of using machine learning methodology to mine information from PCP data concerning protein sequences, as well as the superiority of EBMC, SVM and LR (especially EBMC) for the ubiquitination prediction compared to other methods.
泛素化是蛋白质翻译后修饰中一个非常重要的过程,生物科学家和研究人员对此进行了广泛研究。已经开发出不同的实验和计算方法来识别蛋白质序列中的泛素化位点。本文旨在探索利用蛋白质序列中氨基酸的物理化学性质(PCP)进行泛素化位点预测的计算机学习方法。
我们首先建立了六个不同的泛素化数据集,其记录包含不同数量蛋白质序列片段中的泛素化位点和非泛素化位点。具体而言,为建立这些数据集,从四篇已发表的关于泛素化的论文中使用的原始蛋白质序列中提取蛋白质序列片段,同时基于来自AAindex(氨基酸索引数据库)的PCP值,通过对每个片段上所有氨基酸的PCP值求平均值,计算每个提取的蛋白质序列片段的531个PCP特征。然后,将各种计算机学习方法,包括四种贝叶斯网络方法(即朴素贝叶斯(NB)、特征选择NB(FSNB)、模型平均NB(MANB)和高效贝叶斯多变量分类器(EBMC))以及三种回归方法(即支持向量机(SVM)、逻辑回归(LR)和最小绝对收缩和选择算子(LASSO))应用于六个已建立的片段-PCP数据集。采用五折交叉验证和受试者工作特征曲线下面积(AUROC)来评估每种方法的泛素化预测性能。结果表明,蛋白质序列的PCP数据包含可被机器学习方法挖掘用于泛素化位点预测的信息。比较结果表明,EBMC、SVM和LR的性能优于其他方法,并且EBMC是唯一一种对于六个已建立的数据集能够获得大于或等于0.6的AUC值的方法。结果还表明,对于更大的数据,EBMC往往表现得更好。
基于蛋白质序列中氨基酸的物理化学性质,采用机器学习方法进行泛素化位点预测。结果证明了使用机器学习方法从关于蛋白质序列的PCP数据中挖掘信息的有效性,以及与其他方法相比,EBMC、SVM和LR(尤其是EBMC)在泛素化预测方面的优越性。