Jacobs Matthieu, Grégoire Nicolas, Couet William, Bulitta Jurgen B
INSERM U1070, Poitiers, France.
Center for Pharmacometrics and Systems Pharmacology, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida, United States of America.
PLoS Comput Biol. 2016 Mar 11;12(3):e1004782. doi: 10.1371/journal.pcbi.1004782. eCollection 2016 Mar.
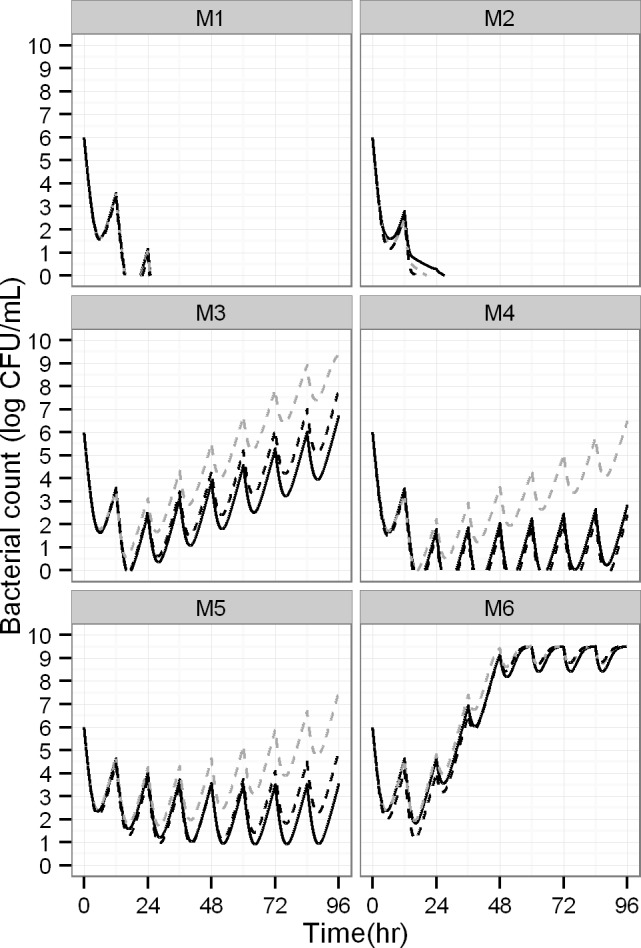
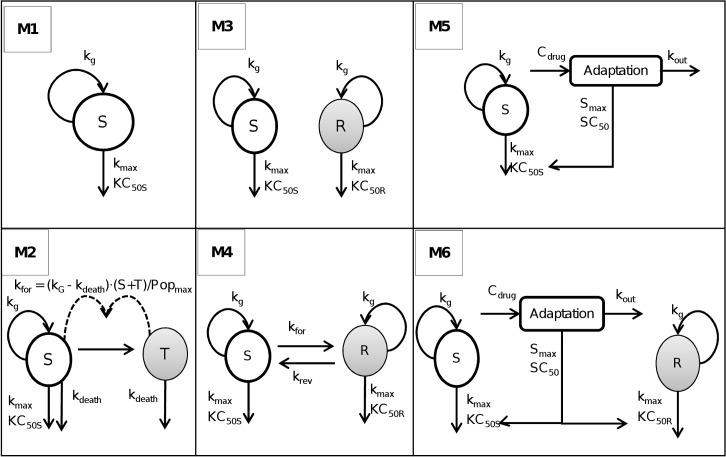
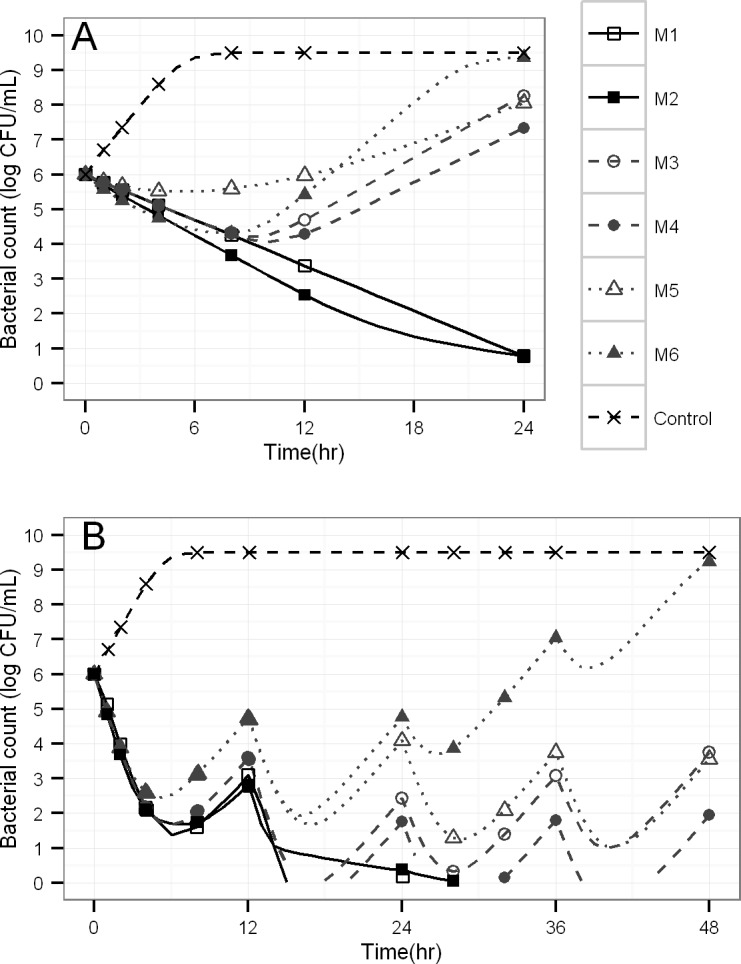
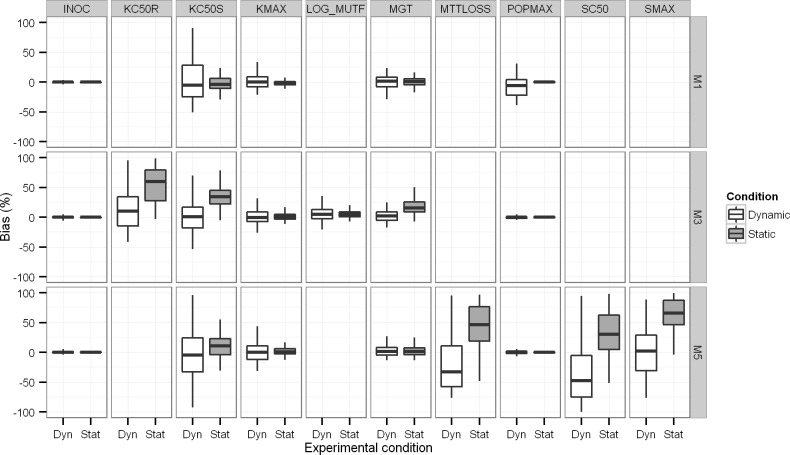
Semi-mechanistic pharmacokinetic-pharmacodynamic (PK-PD) modeling is increasingly used for antimicrobial drug development and optimization of dosage regimens, but systematic simulation-estimation studies to distinguish between competing PD models are lacking. This study compared the ability of static and dynamic in vitro infection models to distinguish between models with different resistance mechanisms and support accurate and precise parameter estimation. Monte Carlo simulations (MCS) were performed for models with one susceptible bacterial population without (M1) or with a resting stage (M2), a one population model with adaptive resistance (M5), models with pre-existing susceptible and resistant populations without (M3) or with (M4) inter-conversion, and a model with two pre-existing populations with adaptive resistance (M6). For each model, 200 datasets of the total bacterial population were simulated over 24h using static antibiotic concentrations (256-fold concentration range) or over 48h under dynamic conditions (dosing every 12h; elimination half-life: 1h). Twelve-hundred random datasets (each containing 20 curves for static or four curves for dynamic conditions) were generated by bootstrapping. Each dataset was estimated by all six models via population PD modeling to compare bias and precision. For M1 and M3, most parameter estimates were unbiased (<10%) and had good imprecision (<30%). However, parameters for adaptive resistance and inter-conversion for M2, M4, M5 and M6 had poor bias and large imprecision under static and dynamic conditions. For datasets that only contained viable counts of the total population, common statistical criteria and diagnostic plots did not support sound identification of the true resistance mechanism. Therefore, it seems advisable to quantify resistant bacteria and characterize their MICs and resistance mechanisms to support extended simulations and translate from in vitro experiments to animal infection models and ultimately patients.
半机制药代动力学-药效学(PK-PD)模型越来越多地用于抗菌药物研发和给药方案优化,但缺乏区分相互竞争的药效学模型的系统模拟-估计研究。本研究比较了静态和动态体外感染模型区分具有不同耐药机制的模型以及支持准确和精确参数估计的能力。对具有一个无静止期(M1)或有静止期(M2)的敏感细菌群体的模型、具有适应性耐药的单群体模型(M5)、具有预先存在的敏感和耐药群体且无(M3)或有(M4)相互转化的模型以及具有两个预先存在的具有适应性耐药群体的模型(M6)进行了蒙特卡罗模拟(MCS)。对于每个模型,使用静态抗生素浓度(256倍浓度范围)在24小时内或在动态条件下(每12小时给药一次;消除半衰期:1小时)在48小时内模拟200个总细菌群体的数据集。通过自抽样生成1200个随机数据集(每个包含20条静态曲线或4条动态条件曲线)。每个数据集通过群体药效学建模由所有六个模型进行估计,以比较偏差和精度。对于M1和M3,大多数参数估计无偏差(<10%)且不精密度良好(<30%)。然而,在静态和动态条件下,M2、M4、M5和M6的适应性耐药和相互转化参数偏差较大且不精密度高。对于仅包含总群体活菌计数的数据集,常用的统计标准和诊断图不支持对真实耐药机制的可靠识别。因此,似乎有必要对抗性细菌进行定量并表征其MIC和耐药机制,以支持扩展模拟并从体外实验转化为动物感染模型并最终应用于患者。