Žurauskienė Justina, Yau Christopher
Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, Oxford, OX3 7BN, UK.
Department of Statistics, University of Oxford, 1 S. Parks Rd, Oxford, OX1 3TG, UK.
BMC Bioinformatics. 2016 Mar 22;17:140. doi: 10.1186/s12859-016-0984-y.
Advances in single cell genomics provide a way of routinely generating transcriptomics data at the single cell level. A frequent requirement of single cell expression analysis is the identification of novel patterns of heterogeneity across single cells that might explain complex cellular states or tissue composition. To date, classical statistical analysis tools have being routinely applied, but there is considerable scope for the development of novel statistical approaches that are better adapted to the challenges of inferring cellular hierarchies.
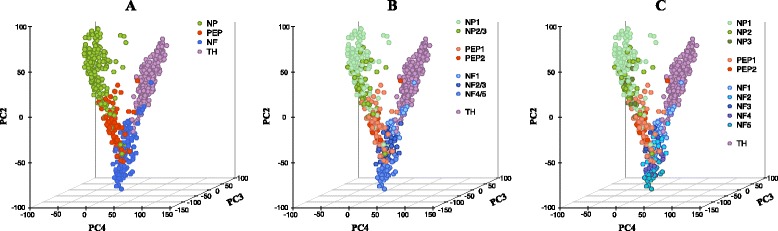
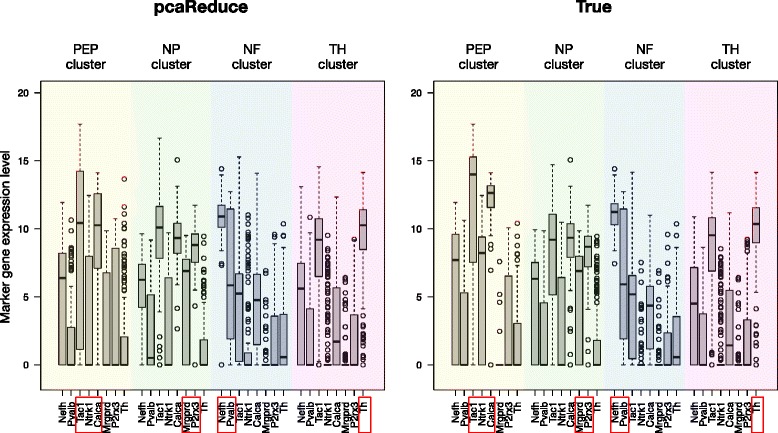
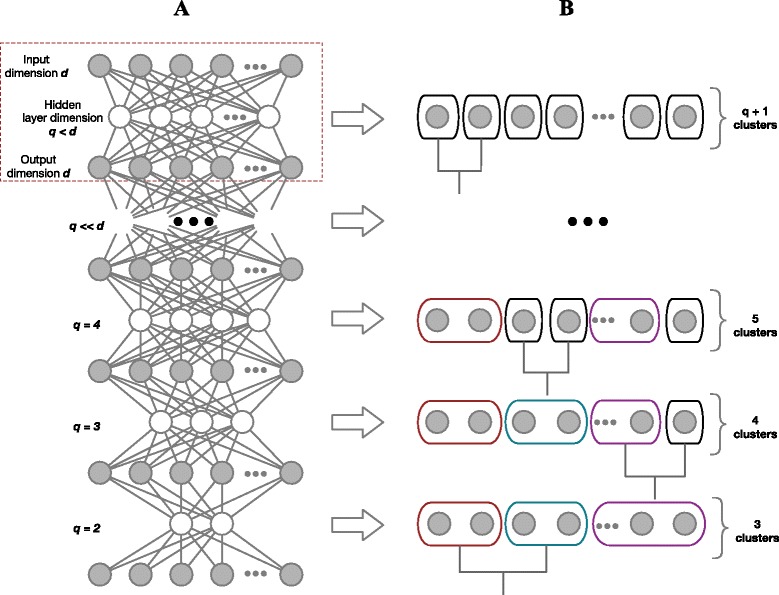
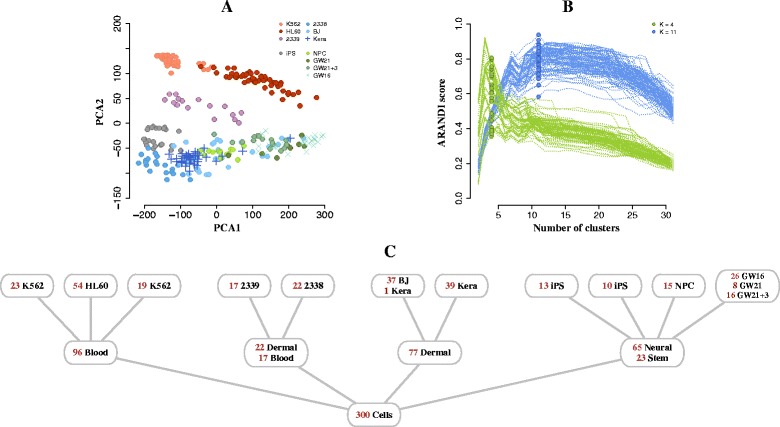
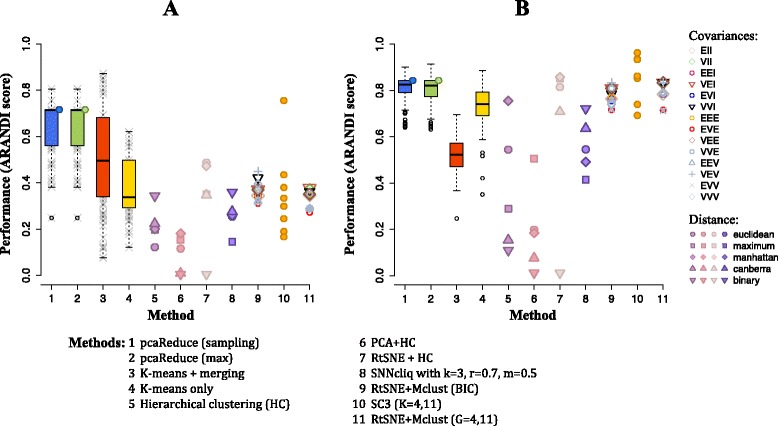
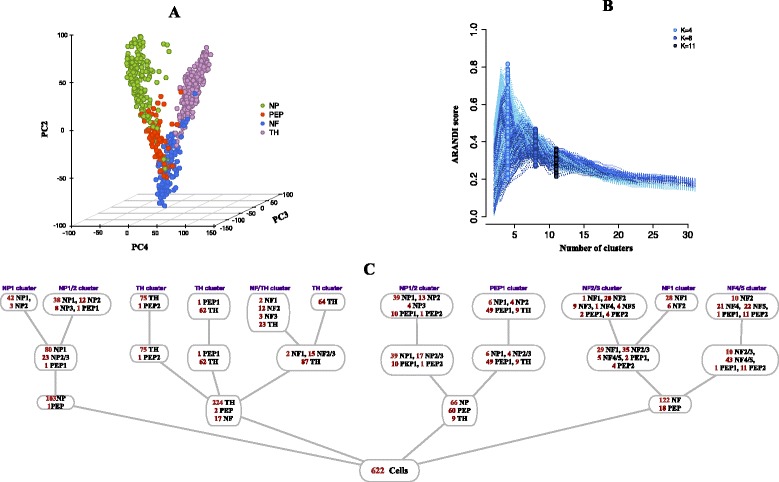
We have developed a novel agglomerative clustering method that we call pcaReduce to generate a cell state hierarchy where each cluster branch is associated with a principal component of variation that can be used to differentiate two cell states. Using two real single cell datasets, we compared our approach to other commonly used statistical techniques, such as K-means and hierarchical clustering. We found that pcaReduce was able to give more consistent clustering structures when compared to broad and detailed cell type labels.
Our novel integration of principal components analysis and hierarchical clustering establishes a connection between the representation of the expression data and the number of cell types that can be discovered. In doing so we found that pcaReduce performs better than either technique in isolation in terms of characterising putative cell states. Our methodology is complimentary to other single cell clustering techniques and adds to a growing palette of single cell bioinformatics tools for profiling heterogeneous cell populations.
单细胞基因组学的进展提供了一种在单细胞水平上常规生成转录组学数据的方法。单细胞表达分析的一个常见要求是识别单细胞间新的异质性模式,这些模式可能解释复杂的细胞状态或组织组成。迄今为止,经典统计分析工具已被常规应用,但开发更适合推断细胞层次结构挑战的新型统计方法仍有很大空间。
我们开发了一种新颖的凝聚聚类方法,称为pcaReduce,以生成细胞状态层次结构,其中每个聚类分支与可用于区分两种细胞状态的主要变异成分相关联。使用两个真实的单细胞数据集,我们将我们的方法与其他常用统计技术(如K均值和层次聚类)进行了比较。我们发现,与宽泛和详细的细胞类型标签相比,pcaReduce能够给出更一致的聚类结构。
我们对主成分分析和层次聚类的新颖整合在表达数据的表示与可发现的细胞类型数量之间建立了联系。通过这样做,我们发现pcaReduce在表征假定的细胞状态方面比单独使用任何一种技术都表现得更好。我们的方法是对其他单细胞聚类技术的补充,并为分析异质细胞群体的单细胞生物信息学工具增添了新的内容。