Branchu Julien, Boutry Maxime, Sourd Laura, Depp Marine, Leone Céline, Corriger Alexandrine, Vallucci Maeva, Esteves Typhaine, Matusiak Raphaël, Dumont Magali, Muriel Marie-Paule, Santorelli Filippo M, Brice Alexis, El Hachimi Khalid Hamid, Stevanin Giovanni, Darios Frédéric
Sorbonne Universités, UPMC Univ Paris 06, UMR S 1127, F-75013 Paris, France; Inserm, U1127, F-75013 Paris, France; CNRS, UMR 7225, F-75013 Paris, France; Institut du Cerveau et de la Moelle épinière, ICM, F-75013 Paris, France.
Sorbonne Universités, UPMC Univ Paris 06, UMR S 1127, F-75013 Paris, France; Inserm, U1127, F-75013 Paris, France; CNRS, UMR 7225, F-75013 Paris, France; Institut du Cerveau et de la Moelle épinière, ICM, F-75013 Paris, France; Ecole Pratique des Hautes Etudes, PSL Research University, Laboratoire de Neurogénétique, F-75013 Paris, France.
Neurobiol Dis. 2017 Jun;102:21-37. doi: 10.1016/j.nbd.2017.02.007. Epub 2017 Feb 22.
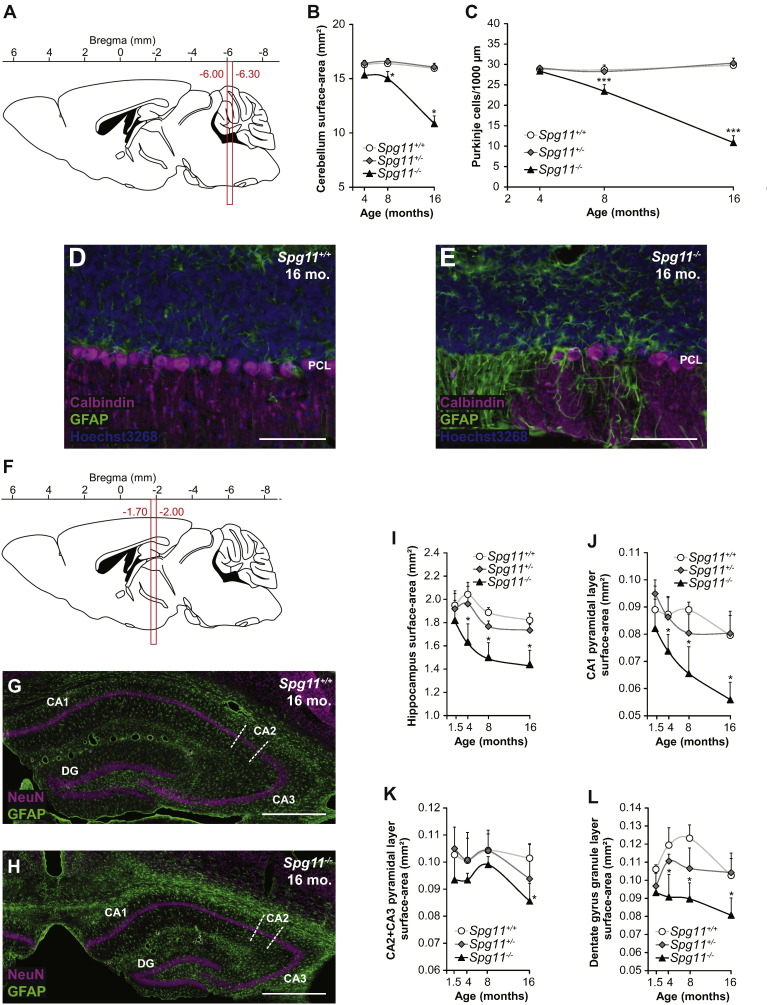
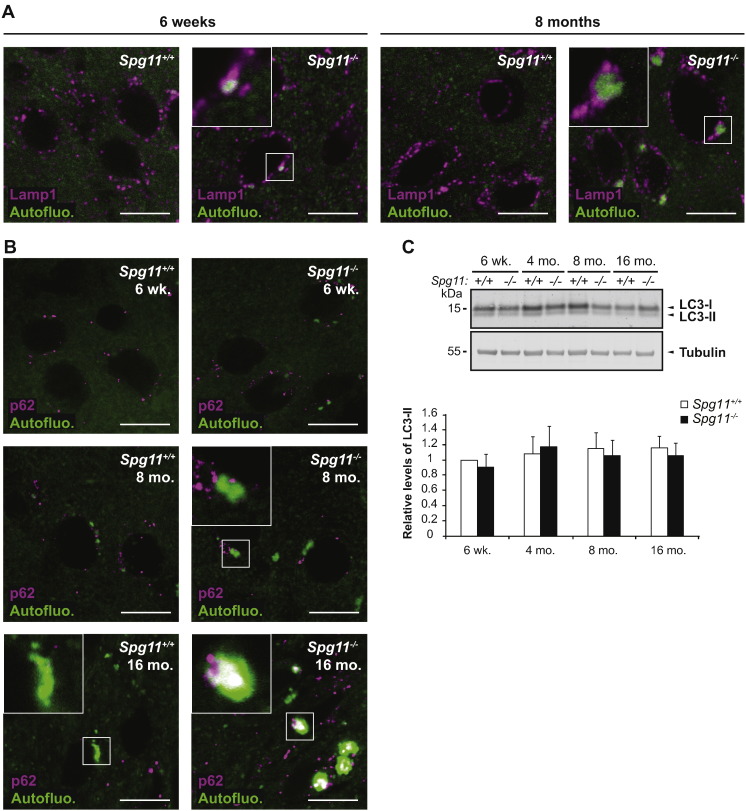
Mutations in SPG11 account for the most common form of autosomal recessive hereditary spastic paraplegia (HSP), characterized by a gait disorder associated with various brain alterations. Mutations in the same gene are also responsible for rare forms of Charcot-Marie-Tooth (CMT) disease and progressive juvenile-onset amyotrophic lateral sclerosis (ALS). To elucidate the physiopathological mechanisms underlying these human pathologies, we disrupted the Spg11 gene in mice by inserting stop codons in exon 32, mimicking the most frequent mutations found in patients. The Spg11 knockout mouse developed early-onset motor impairment and cognitive deficits. These behavioral deficits were associated with progressive brain atrophy with the loss of neurons in the primary motor cortex, cerebellum and hippocampus, as well as with accumulation of dystrophic axons in the corticospinal tract. Spinal motor neurons also degenerated and this was accompanied by fragmentation of neuromuscular junctions and muscle atrophy. This new Spg11 knockout mouse therefore recapitulates the full range of symptoms associated with SPG11 mutations observed in HSP, ALS and CMT patients. Examination of the cellular alterations observed in this model suggests that the loss of spatacsin leads to the accumulation of lipids in lysosomes by perturbing their clearance from these organelles. Altogether, our results link lysosomal dysfunction and lipid metabolism to neurodegeneration and pinpoint a critical role of spatacsin in lipid turnover.
SPG11基因的突变是常染色体隐性遗传性痉挛性截瘫(HSP)最常见的病因,其特征为步态障碍并伴有各种脑部病变。同一基因的突变也是罕见型夏科-马里-图斯病(CMT)和进行性青少年型肌萎缩侧索硬化症(ALS)的病因。为了阐明这些人类疾病背后的生理病理机制,我们通过在第32外显子中插入终止密码子来破坏小鼠的Spg11基因,模拟患者中发现的最常见突变。Spg11基因敲除小鼠出现了早发性运动障碍和认知缺陷。这些行为缺陷与渐进性脑萎缩有关,伴有初级运动皮层、小脑和海马体中的神经元丧失,以及皮质脊髓束中营养不良轴突的积累。脊髓运动神经元也发生退化,并伴有神经肌肉接头的断裂和肌肉萎缩。因此,这种新的Spg11基因敲除小鼠概括了在HSP、ALS和CMT患者中观察到的与SPG11突变相关的所有症状。对该模型中观察到的细胞变化的检查表明,spatacsin的缺失通过干扰溶酶体中脂质的清除而导致脂质在溶酶体中积累。总之,我们的结果将溶酶体功能障碍和脂质代谢与神经退行性变联系起来,并确定了spatacsin在脂质周转中的关键作用。