Nielsen Morten, Andreatta Massimo
Instituto de Investigaciones Biotecnológicas, Universidad Nacional de San Martín, Buenos Aires, Argentina.
Center for Biological Sequence Analysis, Technical University of Denmark, Kgs. Lyngby, Denmark.
Genome Med. 2016 Mar 30;8(1):33. doi: 10.1186/s13073-016-0288-x.
Binding of peptides to MHC class I molecules (MHC-I) is essential for antigen presentation to cytotoxic T-cells.
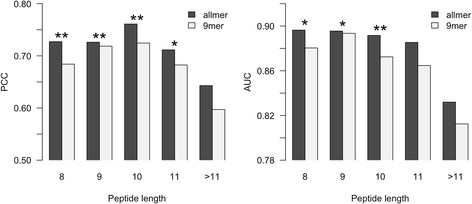
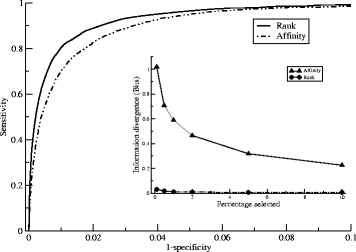
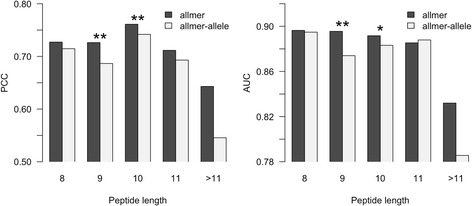
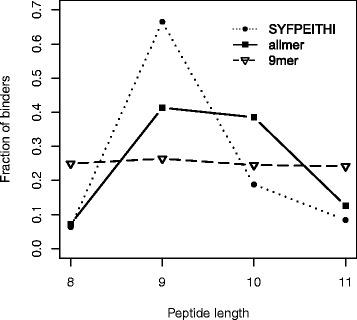
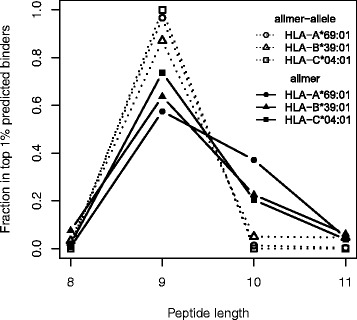
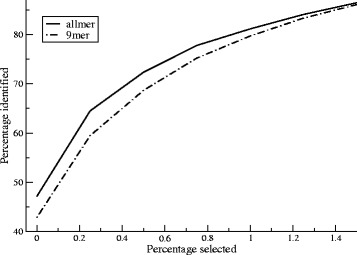
Here, we demonstrate how a simple alignment step allowing insertions and deletions in a pan-specific MHC-I binding machine-learning model enables combining information across both multiple MHC molecules and peptide lengths. This pan-allele/pan-length algorithm significantly outperforms state-of-the-art methods, and captures differences in the length profile of binders to different MHC molecules leading to increased accuracy for ligand identification. Using this model, we demonstrate that percentile ranks in contrast to affinity-based thresholds are optimal for ligand identification due to uniform sampling of the MHC space.
We have developed a neural network-based machine-learning algorithm leveraging information across multiple receptor specificities and ligand length scales, and demonstrated how this approach significantly improves the accuracy for prediction of peptide binding and identification of MHC ligands. The method is available at www.cbs.dtu.dk/services/NetMHCpan-3.0 .
肽与MHC I类分子(MHC-I)的结合对于向细胞毒性T细胞呈递抗原至关重要。
在此,我们展示了在泛特异性MHC-I结合机器学习模型中允许插入和缺失的简单比对步骤如何能够跨多个MHC分子和肽长度组合信息。这种泛等位基因/泛长度算法显著优于现有方法,并捕捉到与不同MHC分子结合的配体在长度分布上的差异,从而提高了配体识别的准确性。使用该模型,我们证明,由于对MHC空间的均匀采样,与基于亲和力的阈值相比,百分位数排名对于配体识别是最优的。
我们开发了一种基于神经网络的机器学习算法,利用跨多种受体特异性和配体长度尺度的信息,并证明了这种方法如何显著提高肽结合预测和MHC配体识别的准确性。该方法可在www.cbs.dtu.dk/services/NetMHCpan-3.0获取。