Randhawa Imtiaz A S, Khatkar Mehar S, Thomson Peter C, Raadsma Herman W
Reprogen - Animal Bioscience Group, Faculty of Veterinary Science, The University of Sydney, 425 Werombi Road, Camden, 2570, NSW, Australia.
PLoS One. 2016 Apr 5;11(4):e0153013. doi: 10.1371/journal.pone.0153013. eCollection 2016.
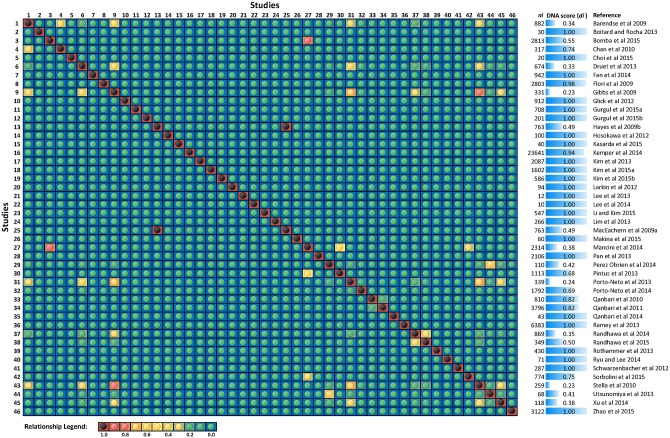
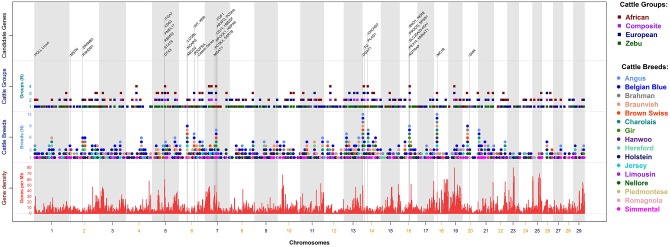
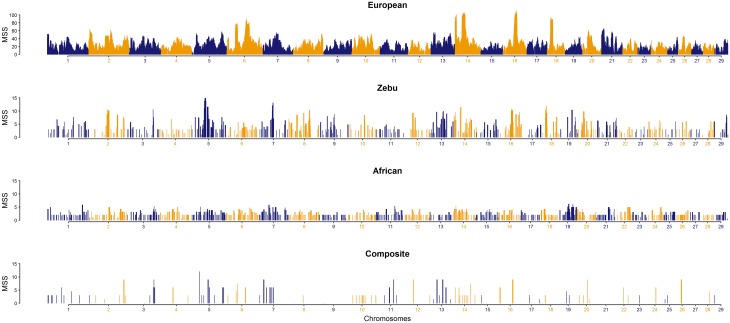
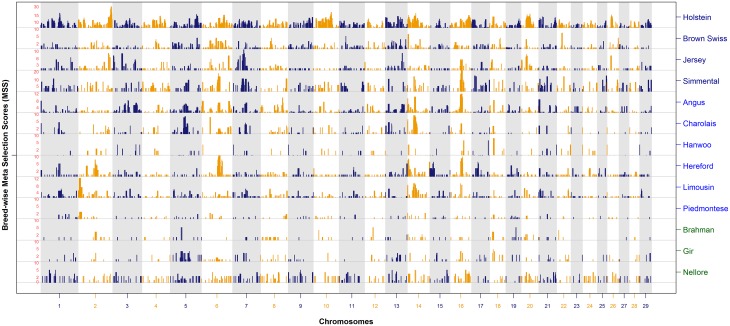
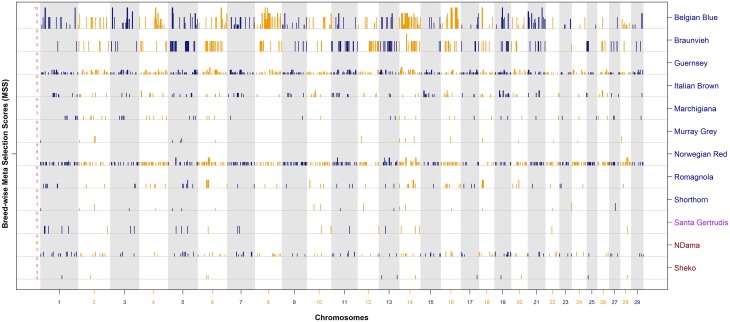
Since domestication, significant genetic improvement has been achieved for many traits of commercial importance in cattle, including adaptation, appearance and production. In response to such intense selection pressures, the bovine genome has undergone changes at the underlying regions of functional genetic variants, which are termed "selection signatures". This article reviews 64 recent (2009-2015) investigations testing genomic diversity for departure from neutrality in worldwide cattle populations. In particular, we constructed a meta-assembly of 16,158 selection signatures for individual breeds and their archetype groups (European, African, Zebu and composite) from 56 genome-wide scans representing 70,743 animals of 90 pure and crossbred cattle breeds. Meta-selection-scores (MSS) were computed by combining published results at every given locus, within a sliding window span. MSS were adjusted for common samples across studies and were weighted for significance thresholds across and within studies. Published selection signatures show extensive coverage across the bovine genome, however, the meta-assembly provides a consensus profile of 263 genomic regions of which 141 were unique (113 were breed-specific) and 122 were shared across cattle archetypes. The most prominent peaks of MSS represent regions under selection across multiple populations and harboured genes of known major effects (coat color, polledness and muscle hypertrophy) and genes known to influence polygenic traits (stature, adaptation, feed efficiency, immunity, behaviour, reproduction, beef and dairy production). As the first meta-assembly of selection signatures, it offers novel insights about the hotspots of selective sweeps in the bovine genome, and this method could equally be applied to other species.
自驯化以来,牛在许多具有商业重要性的性状方面取得了显著的遗传改良,包括适应性、外观和生产性能。为应对如此强烈的选择压力,牛基因组在功能基因变异的潜在区域发生了变化,这些区域被称为“选择印记”。本文综述了最近(2009 - 2015年)64项关于全球牛群中性状偏离中性的基因组多样性测试研究。特别是,我们从代表90个纯系和杂交牛品种的70,743只动物的56次全基因组扫描中,为各个品种及其原型群体(欧洲牛、非洲牛、瘤牛和复合牛)构建了一个包含16,158个选择印记的元组装。通过在滑动窗口范围内合并每个给定基因座的已发表结果来计算元选择分数(MSS)。对跨研究的常见样本进行了MSS调整,并对研究间和研究内的显著性阈值进行了加权。已发表的选择印记显示在牛基因组中广泛覆盖,然而,元组装提供了263个基因组区域的共识图谱,其中141个是独特的(113个是品种特异性的),122个在牛原型群体中共享。MSS最突出的峰值代表多个群体中受到选择的区域,其中包含已知具有主要效应的基因(毛色、无角和肌肉肥大)以及已知影响多基因性状的基因(身高、适应性、饲料效率、免疫力、行为、繁殖、牛肉和奶制品生产)。作为第一个选择印记的元组装,它为牛基因组中选择扫荡的热点提供了新的见解,并且这种方法同样可以应用于其他物种。