Iida Atsushi, Seino Yusuke, Fukami Ayako, Maekawa Ryuya, Yabe Daisuke, Shimizu Shinobu, Kinoshita Keita, Takagi Yusuke, Izumoto Takako, Ogata Hidetada, Ishikawa Kota, Ozaki Nobuaki, Tsunekawa Shin, Hamada Yoji, Oiso Yutaka, Arima Hiroshi, Hayashi Yoshitaka
Department of Endocrinology and Diabetes, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, 4668550, Japan.
Department of Metabolic Medicine, Nagoya University Graduate School of Medicine, Nagoya, Japan.
Diabetologia. 2016 Jul;59(7):1533-1541. doi: 10.1007/s00125-016-3935-2. Epub 2016 Apr 6.
AIMS/HYPOTHESIS: The action of incretin hormones including glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) is potentiated in animal models defective in glucagon action. It has been reported that such animal models maintain normoglycaemia under streptozotocin (STZ)-induced beta cell damage. However, the role of GIP in regulation of glucose metabolism under a combination of glucagon deficiency and STZ-induced beta cell damage has not been fully explored.
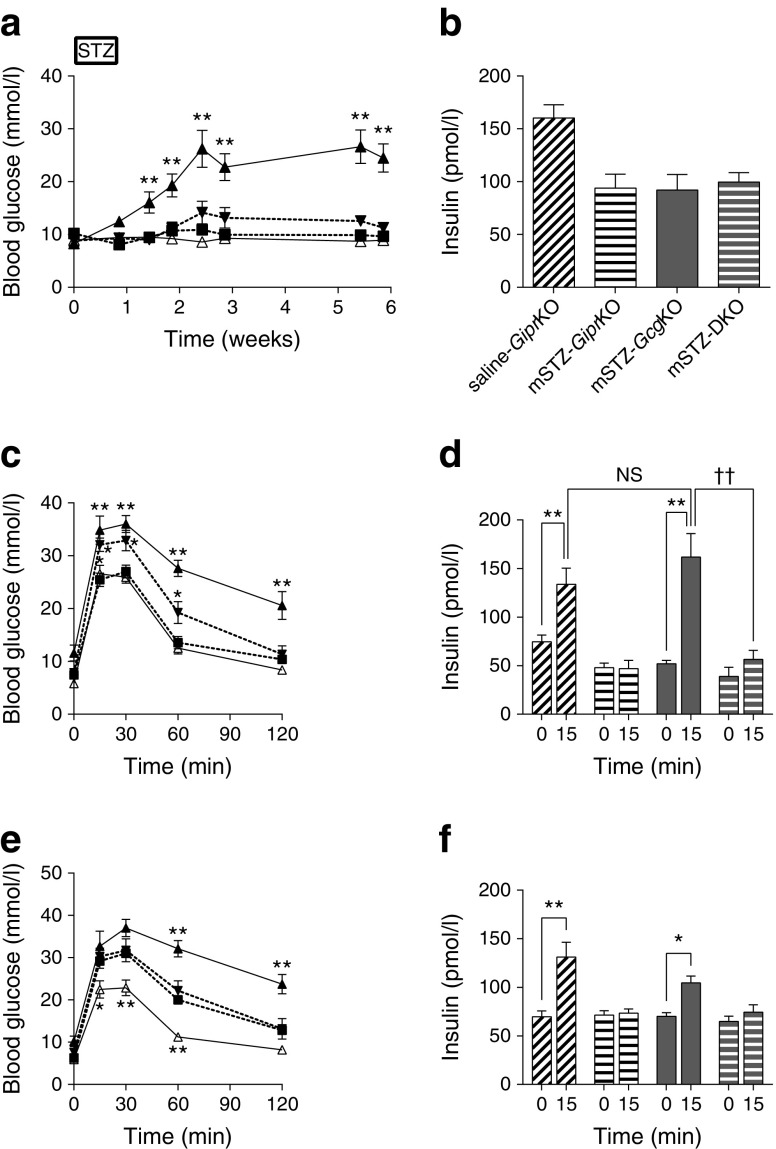
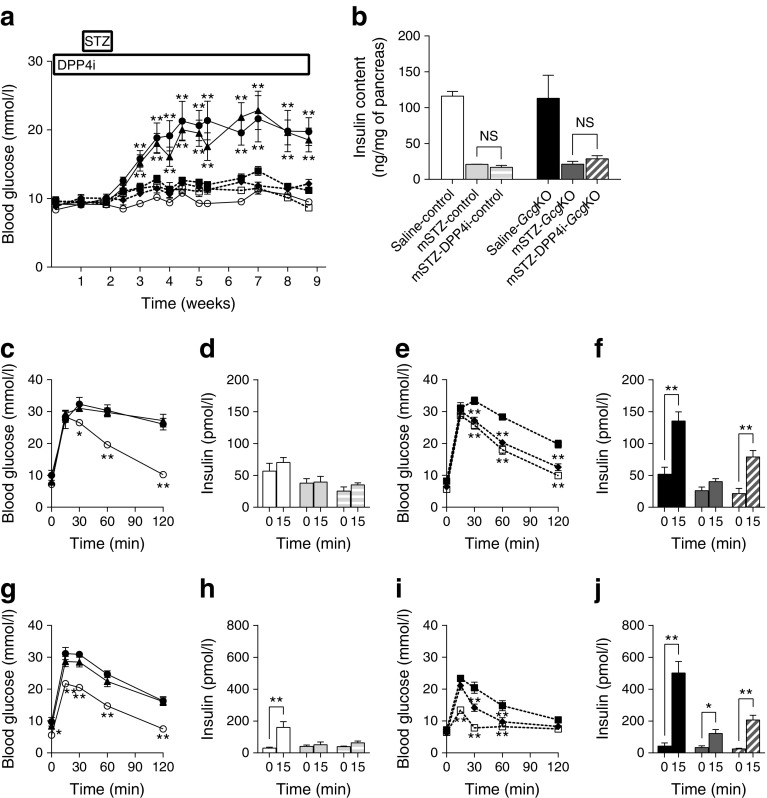
In this study, we investigated glucose metabolism in mice deficient in proglucagon-derived peptides (PGDPs)-namely glucagon gene knockout (GcgKO) mice-administered with STZ. Single high-dose STZ (200 mg/kg, hSTZ) or moderate-dose STZ for five consecutive days (50 mg/kg × 5, mSTZ) was administered to GcgKO mice. The contribution of GIP to glucose metabolism in GcgKO mice was also investigated by experiments employing dipeptidyl peptidase IV (DPP4) inhibitor (DPP4i) or Gcg-Gipr double knockout (DKO) mice.
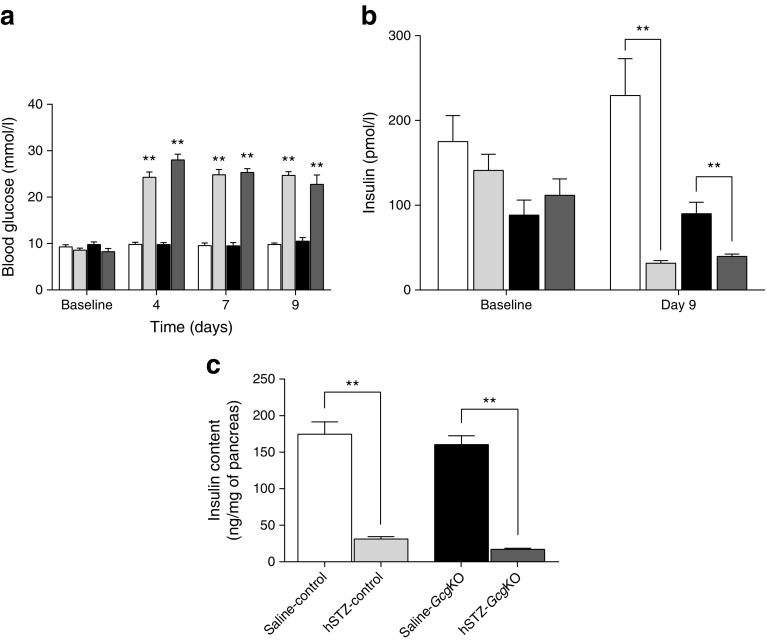
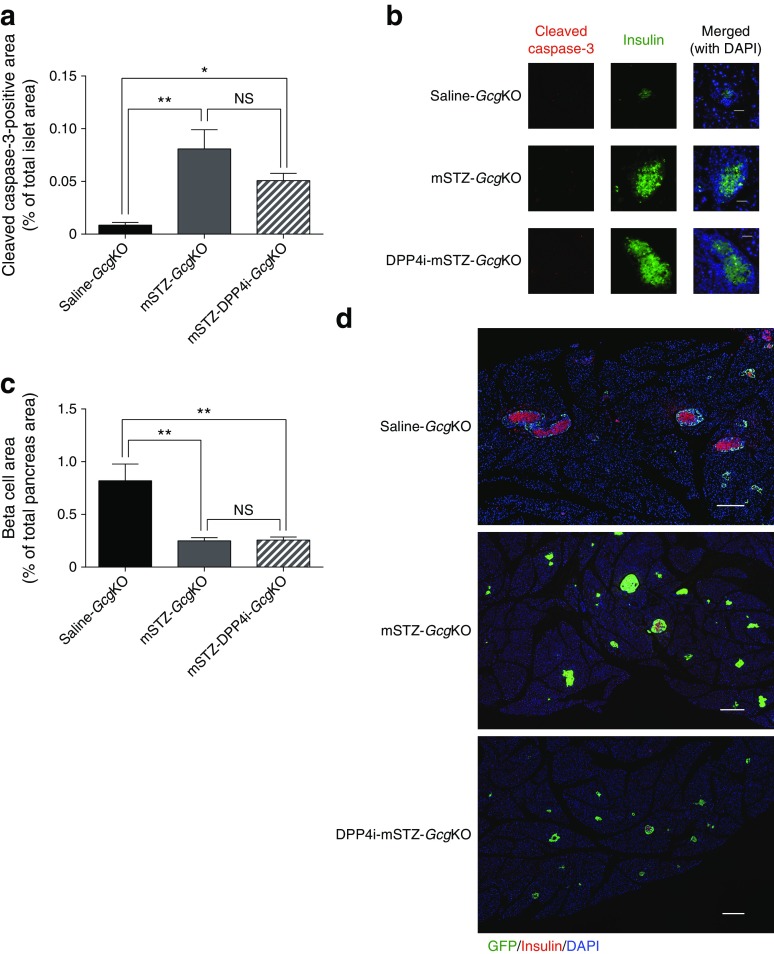
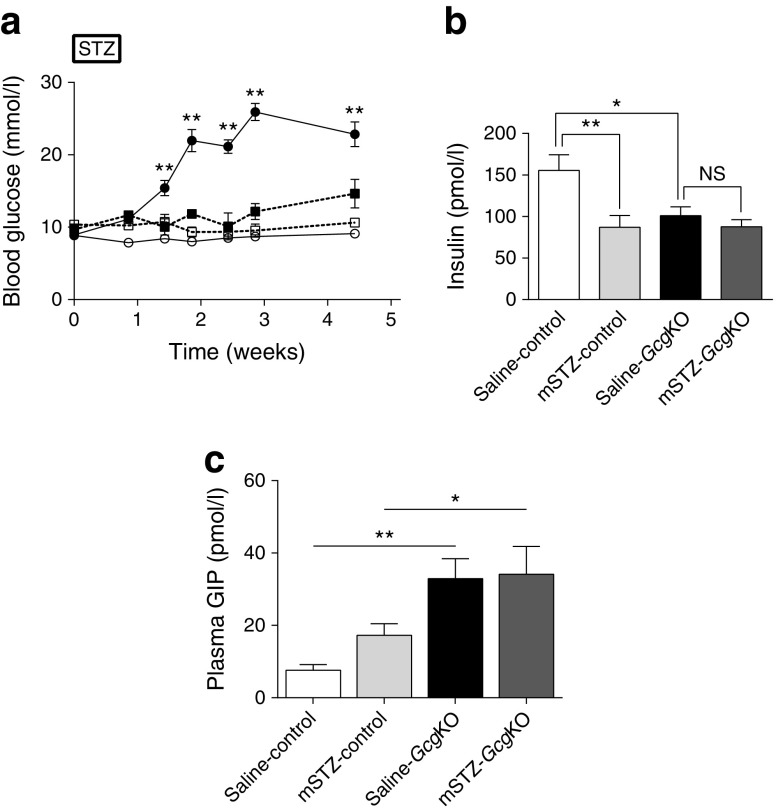
GcgKO mice developed severe diabetes by hSTZ administration despite the absence of glucagon. Administration of mSTZ decreased pancreatic insulin content to 18.8 ± 3.4 (%) in GcgKO mice, but ad libitum-fed blood glucose levels did not significantly increase. Glucose-induced insulin secretion was marginally impaired in mSTZ-treated GcgKO mice but was abolished in mSTZ-treated DKO mice. Although GcgKO mice lack GLP-1, treatment with DPP4i potentiated glucose-induced insulin secretion and ameliorated glucose intolerance in mSTZ-treated GcgKO mice, but did not increase beta cell area or significantly reduce apoptotic cells in islets.
CONCLUSIONS/INTERPRETATION: These results indicate that GIP has the potential to ameliorate glucose intolerance even under STZ-induced beta cell damage by increasing insulin secretion rather than by promoting beta cell survival.
目的/假设:在胰高血糖素作用缺陷的动物模型中,包括葡萄糖依赖性促胰岛素多肽(GIP)和胰高血糖素样肽-1(GLP-1)在内的肠促胰岛素激素的作用会增强。据报道,此类动物模型在链脲佐菌素(STZ)诱导的β细胞损伤下维持正常血糖水平。然而,在胰高血糖素缺乏和STZ诱导的β细胞损伤共同作用下,GIP在葡萄糖代谢调节中的作用尚未得到充分研究。
在本研究中,我们调查了给予STZ的胰高血糖素原衍生肽(PGDPs)缺乏的小鼠(即胰高血糖素基因敲除(GcgKO)小鼠)的葡萄糖代谢情况。对GcgKO小鼠单次给予高剂量STZ(200 mg/kg,hSTZ)或连续五天给予中等剂量STZ(50 mg/kg×5,mSTZ)。还通过使用二肽基肽酶IV(DPP4)抑制剂(DPP4i)或Gcg-Gipr双敲除(DKO)小鼠的实验,研究了GIP对GcgKO小鼠葡萄糖代谢的贡献。
尽管缺乏胰高血糖素,但给予hSTZ后,GcgKO小鼠仍发展为严重糖尿病。给予mSTZ使GcgKO小鼠的胰腺胰岛素含量降至18.8±3.4(%),但自由进食的血糖水平并未显著升高。mSTZ处理的GcgKO小鼠中,葡萄糖诱导的胰岛素分泌略有受损,但在mSTZ处理的DKO小鼠中则被消除。尽管GcgKO小鼠缺乏GLP-1,但用DPP4i治疗可增强mSTZ处理的GcgKO小鼠中葡萄糖诱导的胰岛素分泌,并改善葡萄糖不耐受,但并未增加β细胞面积或显著减少胰岛中的凋亡细胞。
结论/解读:这些结果表明,即使在STZ诱导的β细胞损伤情况下,GIP也有可能通过增加胰岛素分泌而非促进β细胞存活来改善葡萄糖不耐受。