Salvador Ane M, Nevers Tania, Velázquez Francisco, Aronovitz Mark, Wang Bonnie, Abadía Molina Ana, Jaffe Iris Z, Karas Richard H, Blanton Robert M, Alcaide Pilar
Molecular Cardiology Research Institute, Tufts Medical Center, Boston, MA Centro de Investigaciόn Biomédica, Universidad de Granada, Spain.
Molecular Cardiology Research Institute, Tufts Medical Center, Boston, MA.
J Am Heart Assoc. 2016 Mar 15;5(3):e003126. doi: 10.1161/JAHA.115.003126.
Left ventricular dysfunction and heart failure are strongly associated in humans with increased circulating levels of proinflammatory cytokines, T cells, and soluble intercellular cell adhesion molecule 1 (ICAM1). In mice, infiltration of T cells into the left ventricle contributes to pathological cardiac remodeling, but the mechanisms regulating their recruitment to the heart are unclear. We hypothesized that ICAM1 regulates cardiac inflammation and pathological cardiac remodeling by mediating left ventricular T-cell recruitment and thus contributing to cardiac dysfunction and heart failure.
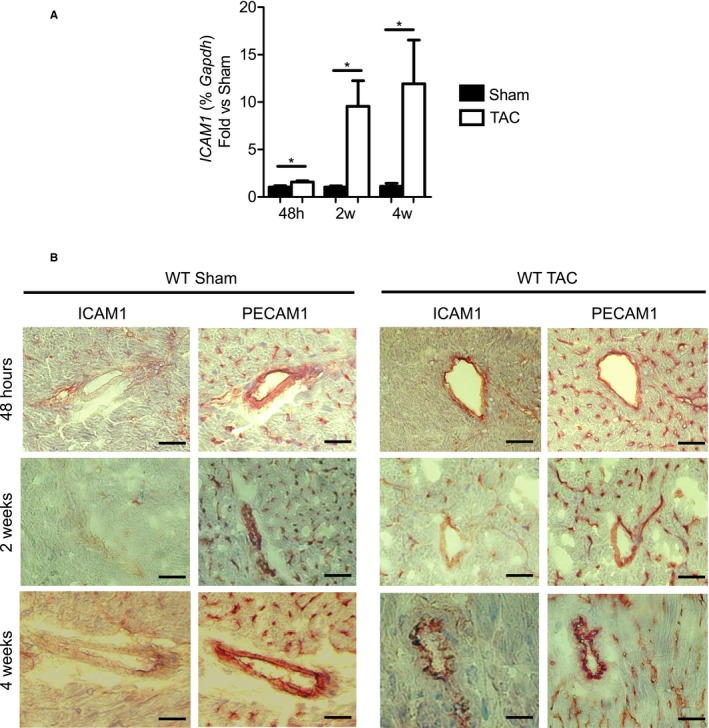
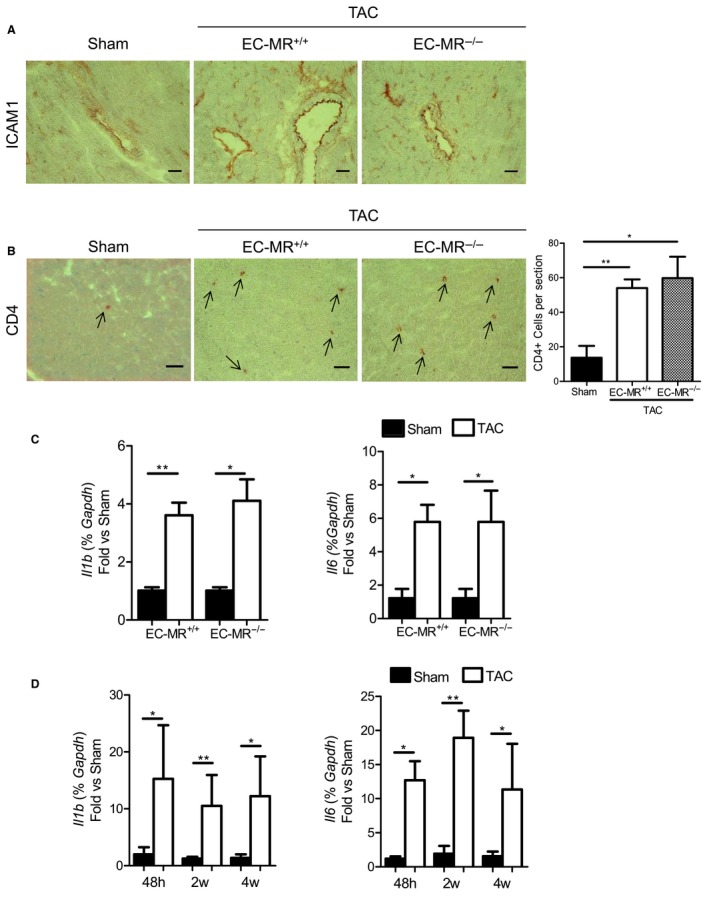
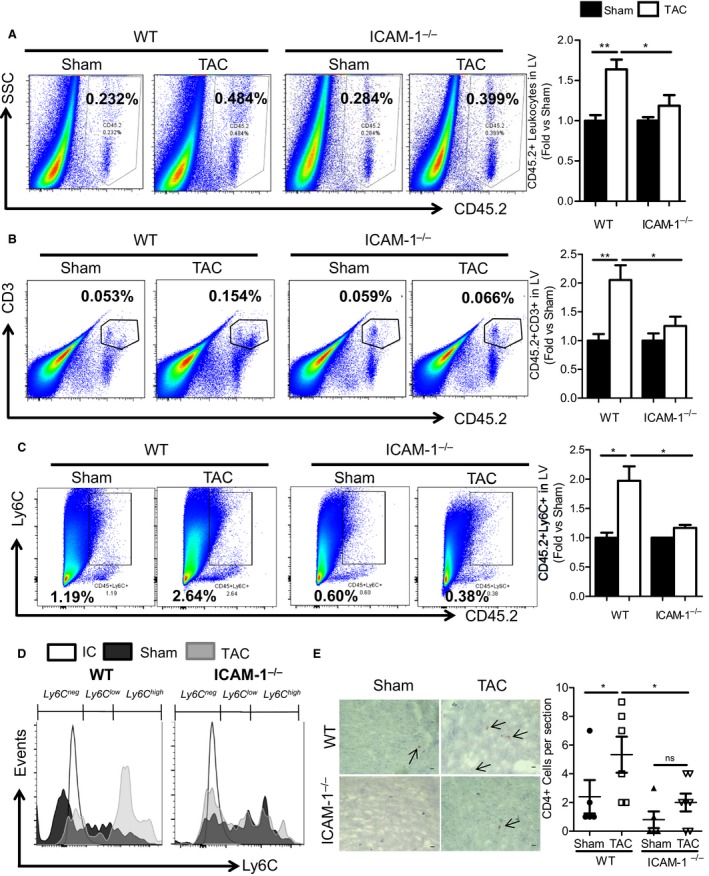
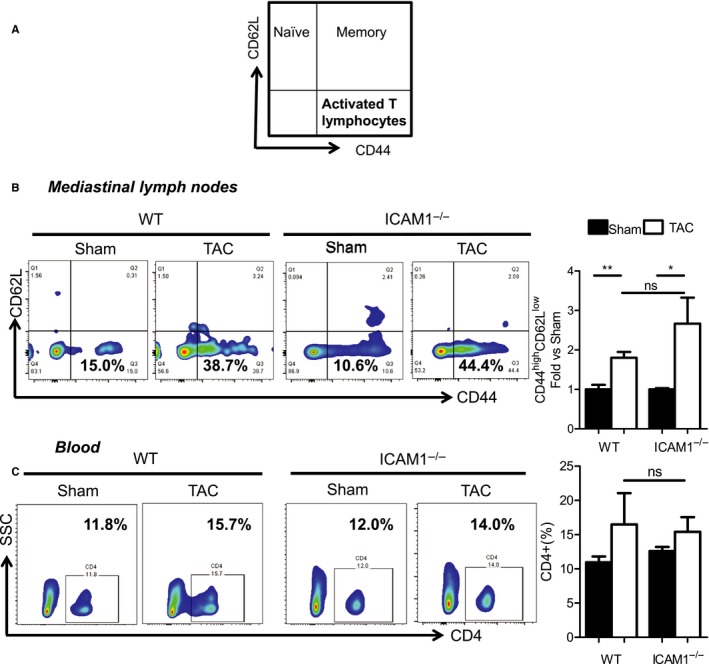
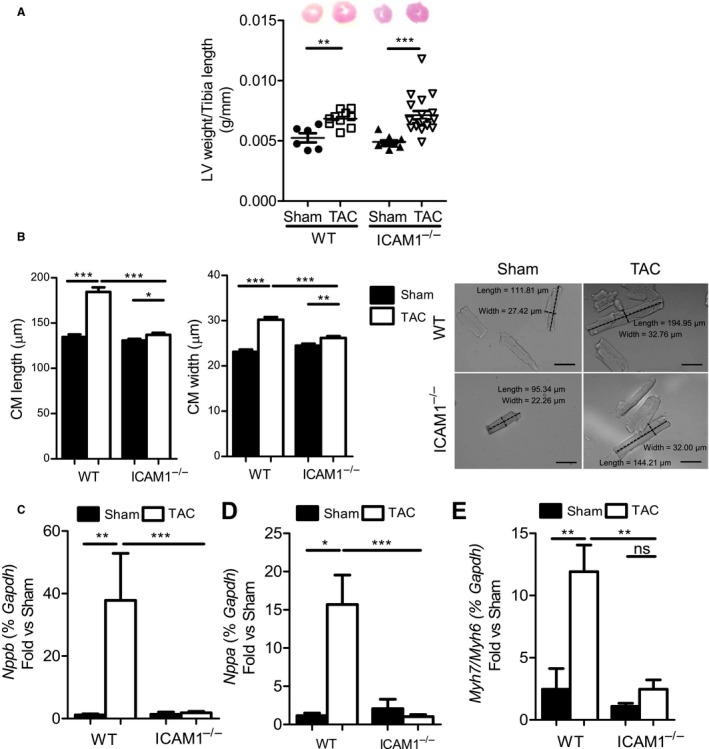
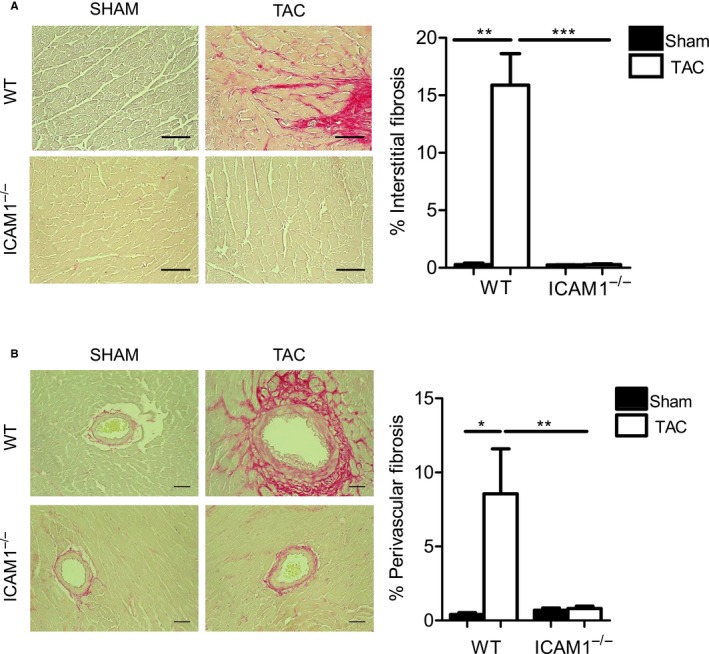
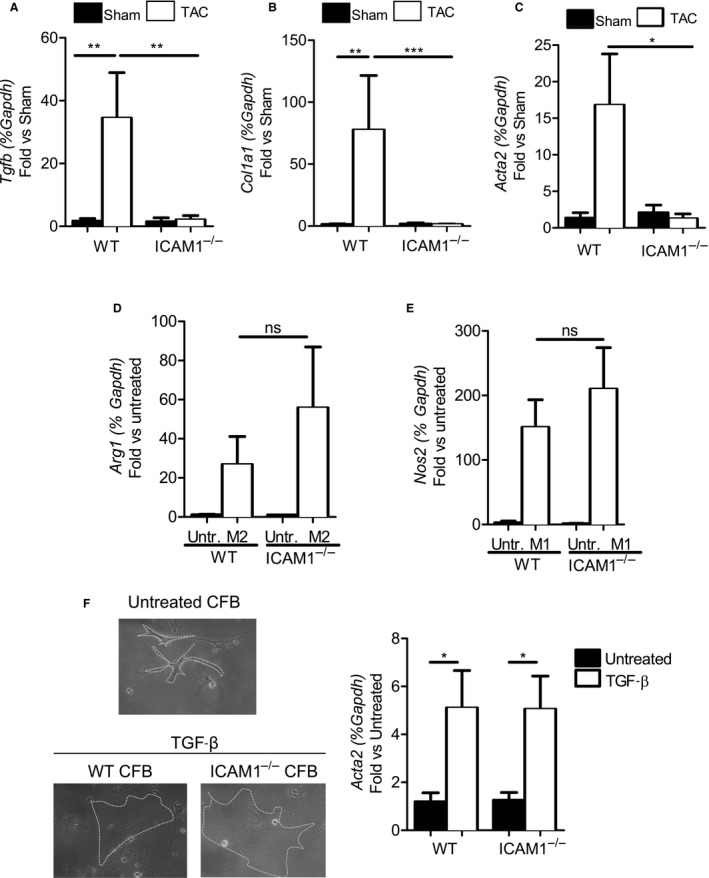
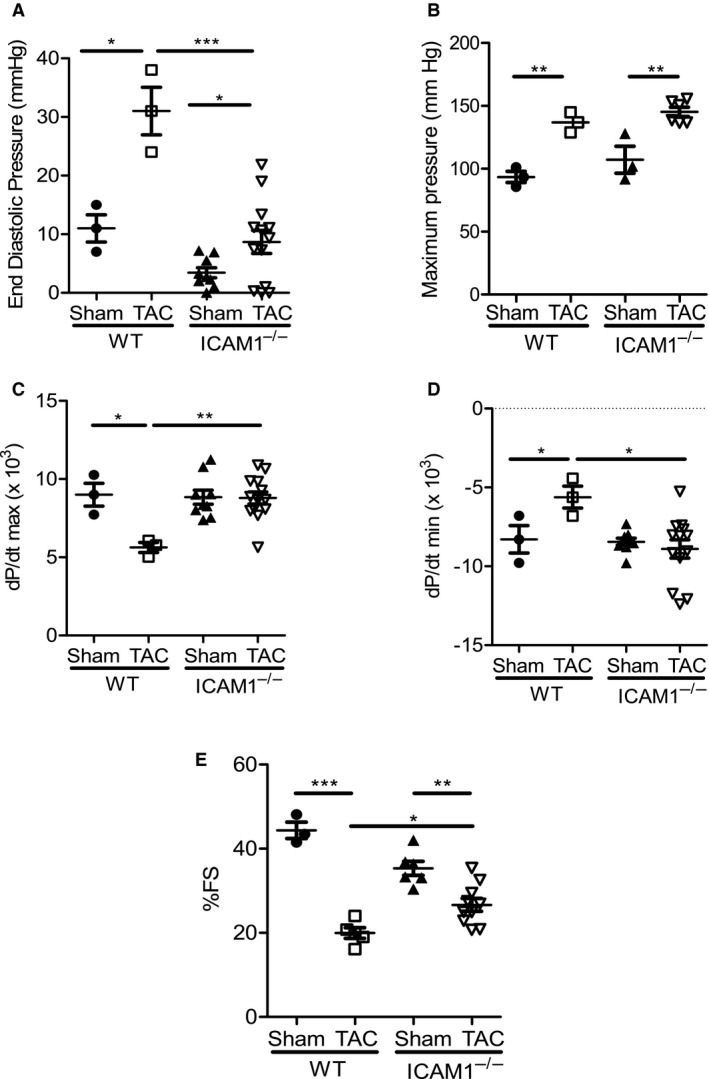
In a mouse model of pressure overload-induced heart failure, intramyocardial endothelial ICAM1 increased within 48 hours in response to thoracic aortic constriction and remained upregulated as heart failure progressed. ICAM1-deficient mice had decreased T-cell and proinflammatory monocyte infiltration in the left ventricle in response to thoracic aortic constriction, despite having numbers of circulating T cells and activated T cells in the heart-draining lymph nodes that were similar to those of wild-type mice. ICAM1-deficient mice did not develop cardiac fibrosis or systolic and diastolic dysfunction in response to thoracic aortic constriction. Exploration of the mechanisms regulating ICAM1 expression revealed that endothelial ICAM1 upregulation and T-cell infiltration were not mediated by endothelial mineralocorticoid receptor signaling, as demonstrated in thoracic aortic constriction studies in mice with endothelial mineralocorticoid receptor deficiency, but rather were induced by the cardiac cytokines interleukin 1β and 6.
ICAM1 regulates pathological cardiac remodeling by mediating proinflammatory leukocyte infiltration in the left ventricle and cardiac fibrosis and dysfunction and thus represents a novel target for treatment of heart failure.
在人类中,左心室功能障碍和心力衰竭与循环中促炎细胞因子、T细胞及可溶性细胞间黏附分子1(ICAM1)水平升高密切相关。在小鼠中,T细胞浸润至左心室会导致病理性心脏重塑,但其募集至心脏的调节机制尚不清楚。我们推测,ICAM1通过介导左心室T细胞募集,进而导致心脏功能障碍和心力衰竭,从而调节心脏炎症和病理性心脏重塑。
在压力超负荷诱导的心力衰竭小鼠模型中,胸主动脉缩窄后48小时内,心肌内内皮ICAM1增加,并随着心力衰竭进展持续上调。尽管ICAM1缺陷小鼠心脏引流淋巴结中的循环T细胞数量和活化T细胞数量与野生型小鼠相似,但胸主动脉缩窄后,其左心室内的T细胞和促炎单核细胞浸润减少。ICAM1缺陷小鼠在胸主动脉缩窄后未出现心脏纤维化或收缩和舒张功能障碍。对调节ICAM1表达机制的探索表明,内皮ICAM1上调和T细胞浸润并非由内皮盐皮质激素受体信号介导,正如内皮盐皮质激素受体缺陷小鼠的胸主动脉缩窄研究所证明的那样,而是由心脏细胞因子白细胞介素1β和6诱导的。
ICAM1通过介导左心室促炎白细胞浸润、心脏纤维化及功能障碍来调节病理性心脏重塑,因此是心力衰竭治疗的一个新靶点。