Jovel Juan, Patterson Jordan, Wang Weiwei, Hotte Naomi, O'Keefe Sandra, Mitchel Troy, Perry Troy, Kao Dina, Mason Andrew L, Madsen Karen L, Wong Gane K-S
Department of Medicine, University of Alberta Edmonton, AB, Canada.
Department of Medicine, University of AlbertaEdmonton, AB, Canada; Department of Biological Sciences, University of AlbertaEdmonton, AB, Canada; BGI-ShenzhenShenzhen, China.
Front Microbiol. 2016 Apr 20;7:459. doi: 10.3389/fmicb.2016.00459. eCollection 2016.
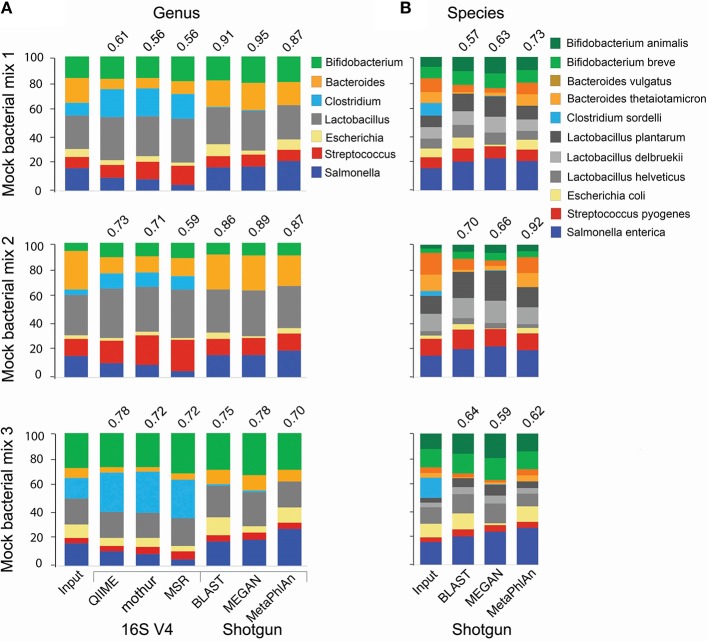
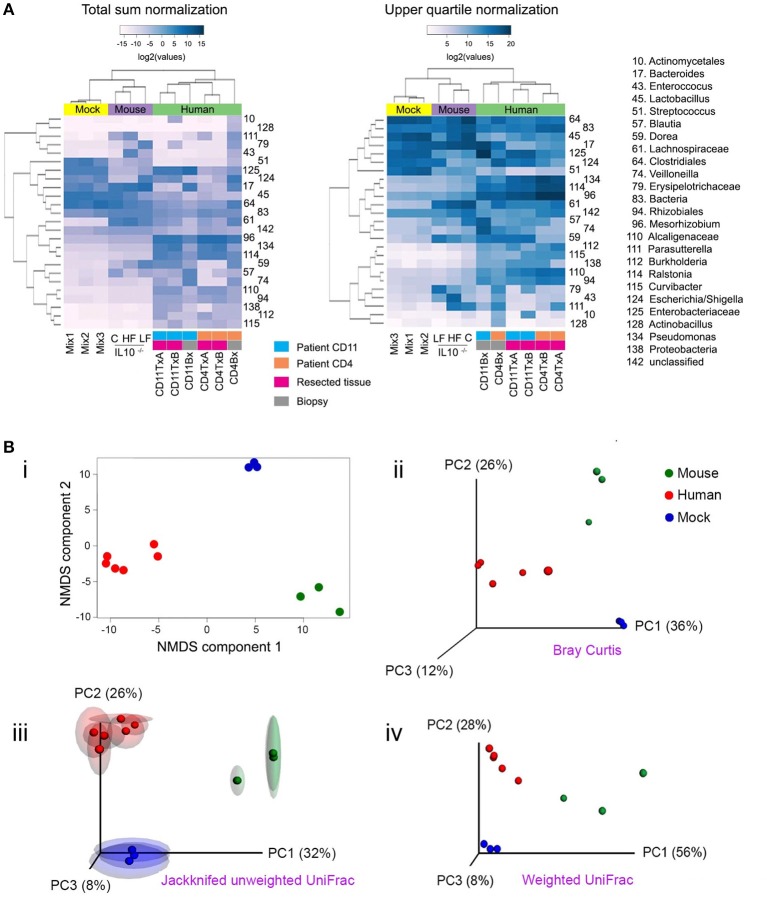
The advent of next generation sequencing (NGS) has enabled investigations of the gut microbiome with unprecedented resolution and throughput. This has stimulated the development of sophisticated bioinformatics tools to analyze the massive amounts of data generated. Researchers therefore need a clear understanding of the key concepts required for the design, execution and interpretation of NGS experiments on microbiomes. We conducted a literature review and used our own data to determine which approaches work best. The two main approaches for analyzing the microbiome, 16S ribosomal RNA (rRNA) gene amplicons and shotgun metagenomics, are illustrated with analyses of libraries designed to highlight their strengths and weaknesses. Several methods for taxonomic classification of bacterial sequences are discussed. We present simulations to assess the number of sequences that are required to perform reliable appraisals of bacterial community structure. To the extent that fluctuations in the diversity of gut bacterial populations correlate with health and disease, we emphasize various techniques for the analysis of bacterial communities within samples (α-diversity) and between samples (β-diversity). Finally, we demonstrate techniques to infer the metabolic capabilities of a bacteria community from these 16S and shotgun data.
新一代测序(NGS)技术的出现,使得对肠道微生物群的研究能够以前所未有的分辨率和通量进行。这推动了复杂生物信息学工具的开发,以分析产生的大量数据。因此,研究人员需要清楚地了解微生物群NGS实验设计、实施和解读所需的关键概念。我们进行了文献综述,并利用我们自己的数据来确定哪种方法效果最佳。通过对旨在突出其优缺点的文库分析,阐述了分析微生物群的两种主要方法,即16S核糖体RNA(rRNA)基因扩增子和鸟枪法宏基因组学。讨论了几种细菌序列分类学分类方法。我们进行了模拟,以评估对细菌群落结构进行可靠评估所需的序列数量。鉴于肠道细菌种群多样性的波动与健康和疾病相关,我们强调了用于分析样本内(α多样性)和样本间(β多样性)细菌群落的各种技术。最后,我们展示了从这些16S和鸟枪法数据推断细菌群落代谢能力的技术。