Hillmann Benjamin, Al-Ghalith Gabriel A, Shields-Cutler Robin R, Zhu Qiyun, Gohl Daryl M, Beckman Kenneth B, Knight Rob, Knights Dan
Department of Computer Science and Engineering, University of Minnesota, Minneapolis, Minnesota, USA.
Bioinformatics and Computational Biology, University of Minnesota, Minneapolis, Minnesota, USA.
mSystems. 2018 Nov 13;3(6). doi: 10.1128/mSystems.00069-18. eCollection 2018 Nov-Dec.
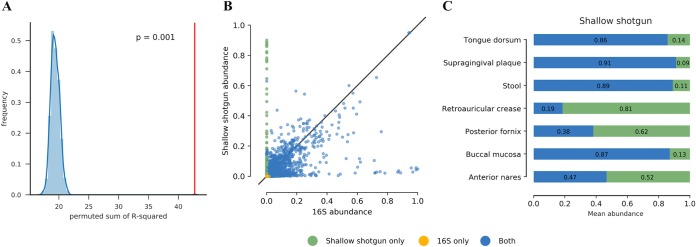
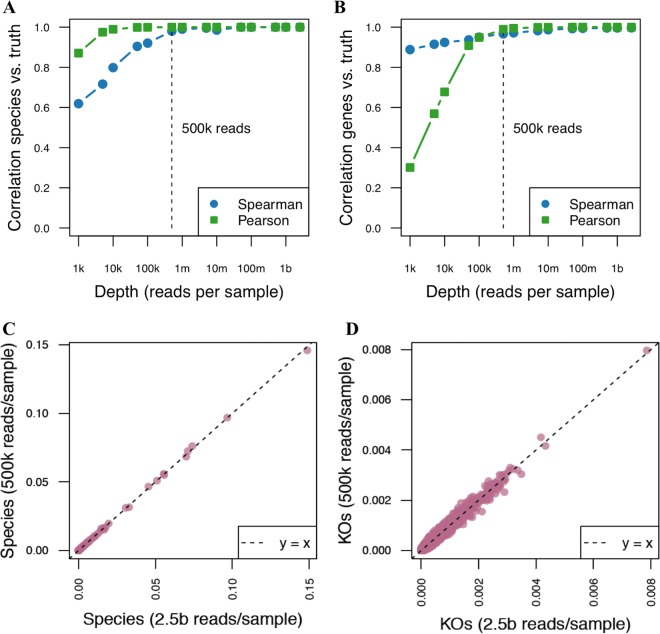
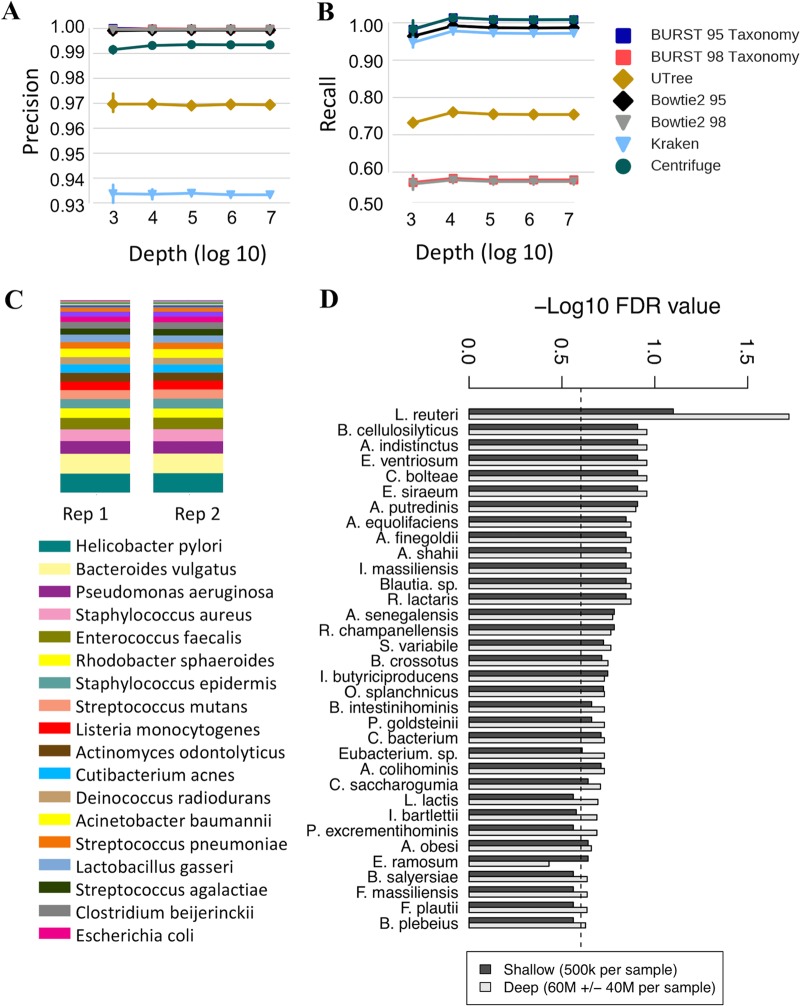
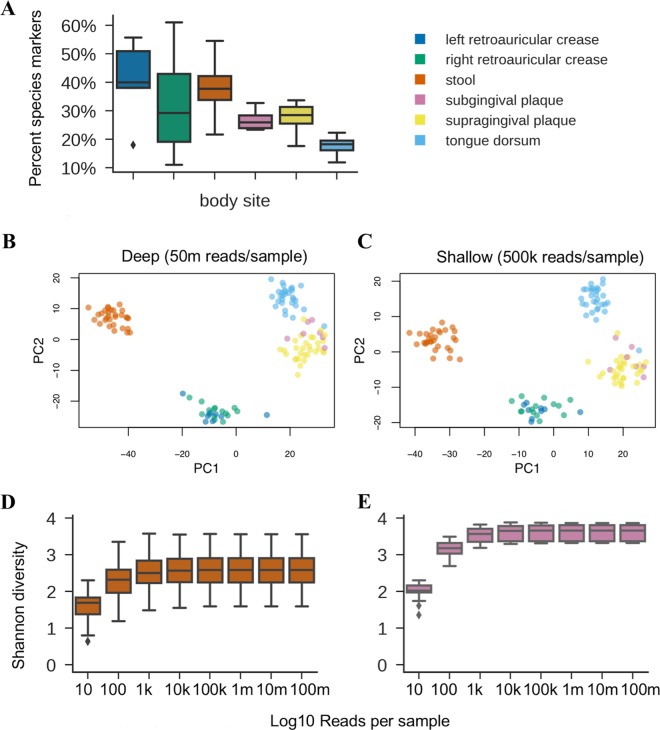
Although microbial communities are associated with human, environmental, plant, and animal health, there exists no cost-effective method for precisely characterizing species and genes in such communities. While deep whole-metagenome shotgun (WMS) sequencing provides high taxonomic and functional resolution, it is often prohibitively expensive for large-scale studies. The prevailing alternative, 16S rRNA gene amplicon (16S) sequencing, often does not resolve taxonomy past the genus level and provides only moderately accurate predictions of the functional profile; thus, there is currently no widely accepted approach to affordable, high-resolution, taxonomic, and functional microbiome analysis. To address this technology gap, we evaluated the information content of shallow shotgun sequencing with as low as 0.5 million sequences per sample as an alternative to 16S sequencing for large human microbiome studies. We describe a library preparation protocol enabling shallow shotgun sequencing at approximately the same per-sample cost as 16S sequencing. We analyzed multiple real and simulated biological data sets, including two novel human stool samples with ultradeep sequencing of 2.5 billion sequences per sample, and found that shallow shotgun sequencing recovers more-accurate species-level taxonomic and functional profiles of the human microbiome than 16S sequencing. We discuss the inherent limitations of shallow shotgun sequencing and note that 16S sequencing remains a valuable and important method for taxonomic profiling of novel environments. Although deep WMS sequencing remains the gold standard for high-resolution microbiome analysis, we recommend that researchers consider shallow shotgun sequencing as a useful alternative to 16S sequencing for large-scale human microbiome research studies where WMS sequencing may be cost-prohibitive. A common refrain in recent microbiome-related academic meetings is that the field needs to move away from broad taxonomic surveys using 16S sequencing and toward more powerful longitudinal studies using shotgun sequencing. However, performing deep shotgun sequencing in large longitudinal studies remains prohibitively expensive for all but the most well-funded research labs and consortia, which leads many researchers to choose 16S sequencing for large studies, followed by deep shotgun sequencing on a subset of targeted samples. Here, we show that shallow- or moderate-depth shotgun sequencing may be used by researchers to obtain species-level taxonomic and functional data at approximately the same cost as amplicon sequencing. While shallow shotgun sequencing is not intended to replace deep shotgun sequencing for strain-level characterization, we recommend that microbiome scientists consider using shallow shotgun sequencing instead of 16S sequencing for large-scale human microbiome studies.
尽管微生物群落与人类、环境、植物和动物健康相关,但目前尚无一种经济高效的方法来精确表征此类群落中的物种和基因。深度全基因组鸟枪法(WMS)测序虽能提供高分辨率的分类学和功能信息,但对于大规模研究而言,其成本往往过高。常用的替代方法——16S rRNA基因扩增子(16S)测序,通常无法在属水平以上解析分类学信息,且对功能谱的预测准确性仅为中等;因此,目前尚无一种被广泛接受的方法可实现经济实惠、高分辨率的分类学和功能微生物组分析。为填补这一技术空白,我们评估了浅度鸟枪法测序的信息含量,该方法每个样本只需低至50万个序列,可作为大规模人类微生物组研究中替代16S测序的方法。我们描述了一种文库制备方案,使浅度鸟枪法测序的每个样本成本与16S测序大致相同。我们分析了多个真实和模拟的生物学数据集,包括两个新型人类粪便样本,每个样本进行了25亿序列的超深度测序,发现浅度鸟枪法测序比16S测序能更准确地恢复人类微生物组的物种水平分类学和功能谱。我们讨论了浅度鸟枪法测序的固有局限性,并指出16S测序对于新环境的分类学分析仍是一种有价值且重要的方法。尽管深度WMS测序仍是高分辨率微生物组分析的金标准,但我们建议研究人员在WMS测序成本过高的大规模人类微生物组研究中,考虑将浅度鸟枪法测序作为16S测序的一种有用替代方法。在近期与微生物组相关的学术会议上,一个常见的观点是,该领域需要从使用16S测序进行广泛的分类学调查,转向使用鸟枪法测序进行更强大的纵向研究。然而,在大型纵向研究中进行深度鸟枪法测序,对于除资金最雄厚的研究实验室和联盟外的所有机构来说,成本仍然过高,这导致许多研究人员在大型研究中选择16S测序,然后对一部分目标样本进行深度鸟枪法测序。在此,我们表明研究人员可以使用浅度或中度深度的鸟枪法测序,以与扩增子测序大致相同的成本获得物种水平的分类学和功能数据。虽然浅度鸟枪法测序并非旨在取代深度鸟枪法测序进行菌株水平的表征,但我们建议微生物组科学家在大规模人类微生物组研究中考虑使用浅度鸟枪法测序而非16S测序。