Moult John, Fidelis Krzysztof, Kryshtafovych Andriy, Schwede Torsten, Tramontano Anna
Institute for Bioscience and Biotechnology Research and Department of Cell Biology and Molecular Genetics, University of Maryland, Rockville, Maryland, 20850.
Genome Center, University of California, Davis, Davis, California, 95616.
Proteins. 2016 Sep;84 Suppl 1(Suppl 1):4-14. doi: 10.1002/prot.25064. Epub 2016 Jun 1.
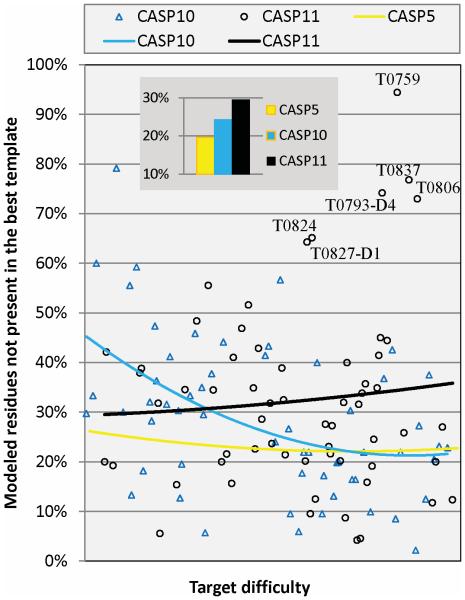
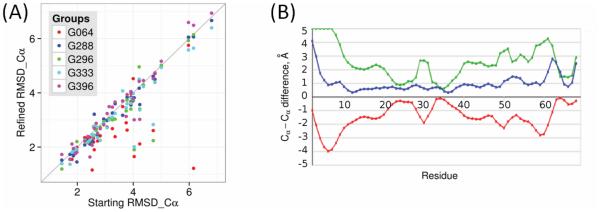
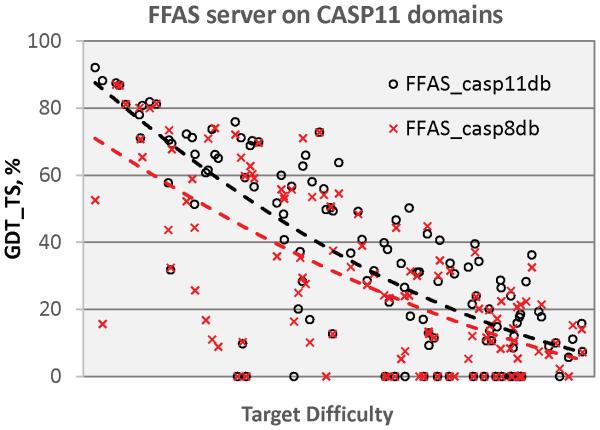
Modeling of protein structure from amino acid sequence now plays a major role in structural biology. Here we report new developments and progress from the CASP11 community experiment, assessing the state of the art in structure modeling. Notable points include the following: (1) New methods for predicting three dimensional contacts resulted in a few spectacular template free models in this CASP, whereas models based on sequence homology to proteins with experimental structure continue to be the most accurate. (2) Refinement of initial protein models, primarily using molecular dynamics related approaches, has now advanced to the point where the best methods can consistently (though slightly) improve nearly all models. (3) The use of relatively sparse NMR constraints dramatically improves the accuracy of models, and another type of sparse data, chemical crosslinking, introduced in this CASP, also shows promise for producing better models. (4) A new emphasis on modeling protein complexes, in collaboration with CAPRI, has produced interesting results, but also shows the need for more focus on this area. (5) Methods for estimating the accuracy of models have advanced to the point where they are of considerable practical use. (6) A first assessment demonstrates that models can sometimes successfully address biological questions that motivate experimental structure determination. (7) There is continuing progress in accuracy of modeling regions of structure not directly available by comparative modeling, while there is marginal or no progress in some other areas. Proteins 2016; 84(Suppl 1):4-14. © 2016 Wiley Periodicals, Inc.
从氨基酸序列预测蛋白质结构的模型构建如今在结构生物学中发挥着重要作用。在此,我们报告了来自CASP11社区实验的新进展,评估了结构建模的当前水平。值得注意的要点如下:(1)预测三维接触的新方法在本次CASP中产生了一些引人注目的无模板模型,而基于与具有实验结构的蛋白质序列同源性的模型仍然是最准确的。(2)初始蛋白质模型的优化,主要使用与分子动力学相关的方法,现已发展到最佳方法能够持续(尽管略有)改进几乎所有模型的程度。(3)使用相对稀疏的核磁共振约束显著提高了模型的准确性,并且在本次CASP中引入的另一种稀疏数据——化学交联,也显示出产生更好模型的前景。(4)与CAPRI合作,对蛋白质复合物建模的新重点产生了有趣的结果,但也表明需要更多地关注这一领域。(5)估计模型准确性的方法已经发展到具有相当实际用途的程度。(6)首次评估表明,模型有时能够成功解决激发实验结构测定的生物学问题。(7)在通过比较建模无法直接获得的结构区域的建模准确性方面持续取得进展,而在其他一些领域则进展甚微或没有进展。《蛋白质》2016年;84(增刊1):4 - 14。©2016威利期刊公司。