Andrews T Daniel, Jeelall Yogesh, Talaulikar Dipti, Goodnow Christopher C, Field Matthew A
Department of Immunology, John Curtin School of Medical Research, Australian National University, Canberra ACT, Australia; National Computational Infrastructure, Canberra ACT, Australia.
Department of Immunology, John Curtin School of Medical Research, Australian National University, Canberra ACT, Australia; School of Medicine and Pharmacology, University of Western Australia, Harry Perkins Institute, Perth, Australia.
PeerJ. 2016 May 24;4:e2074. doi: 10.7717/peerj.2074. eCollection 2016.
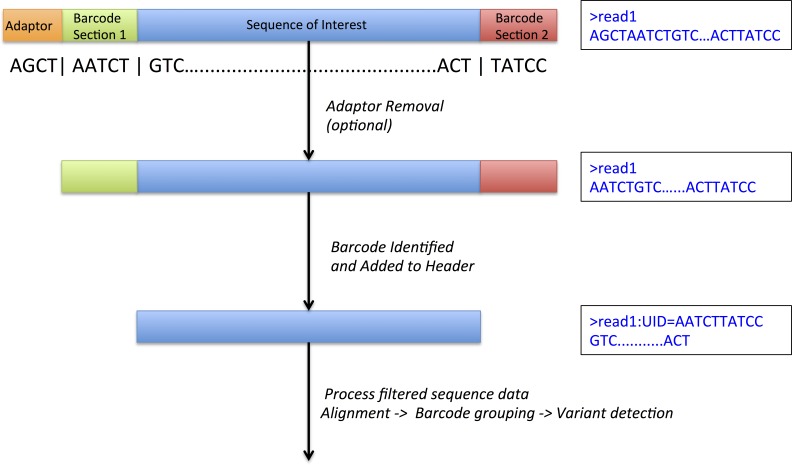
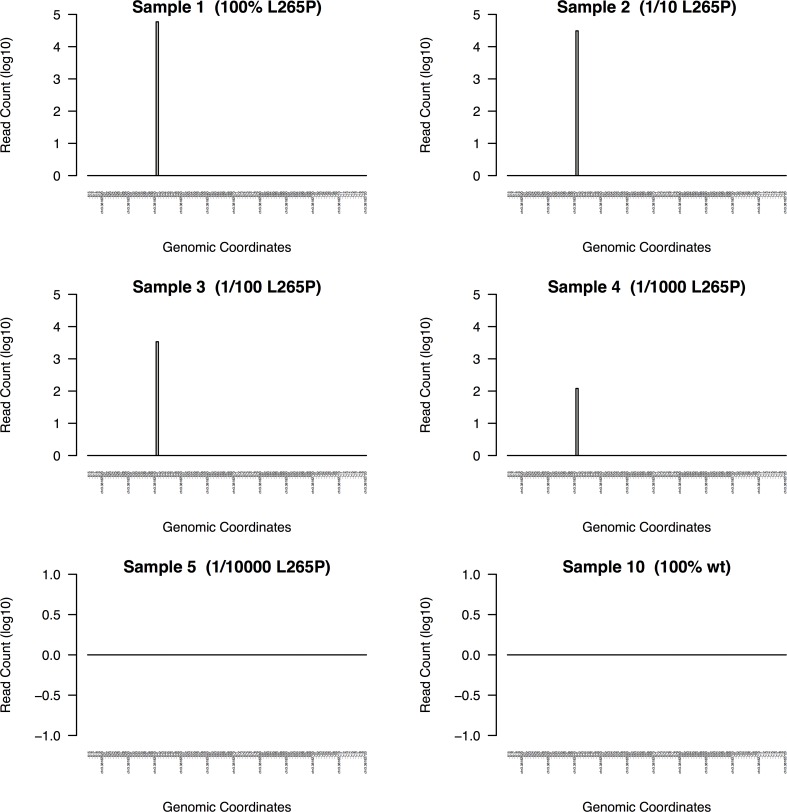
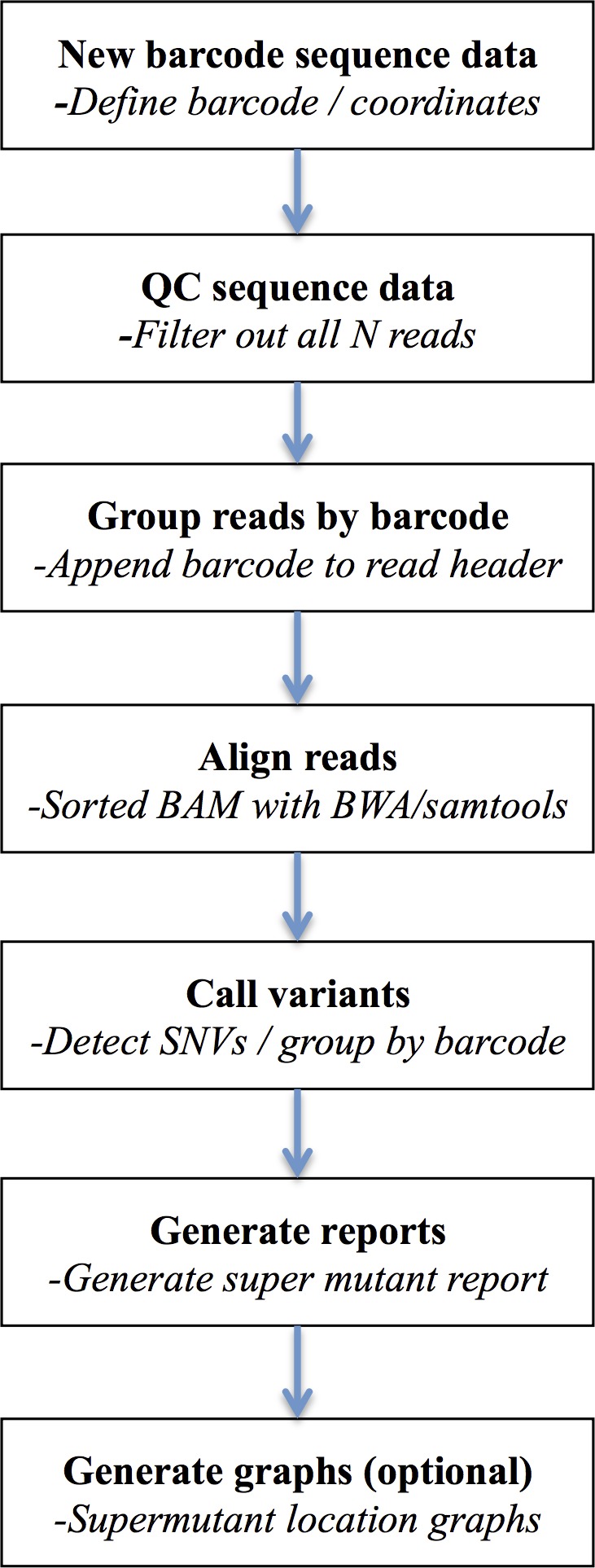
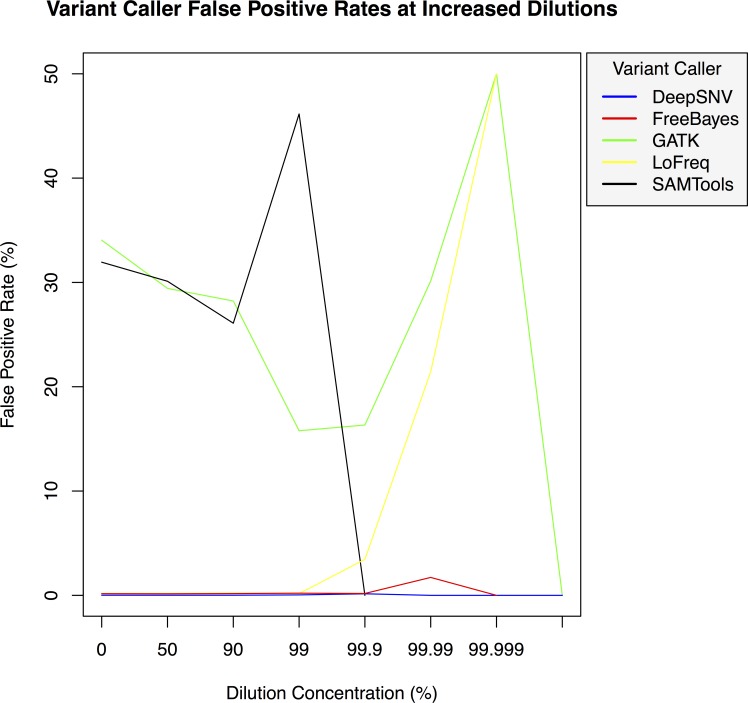
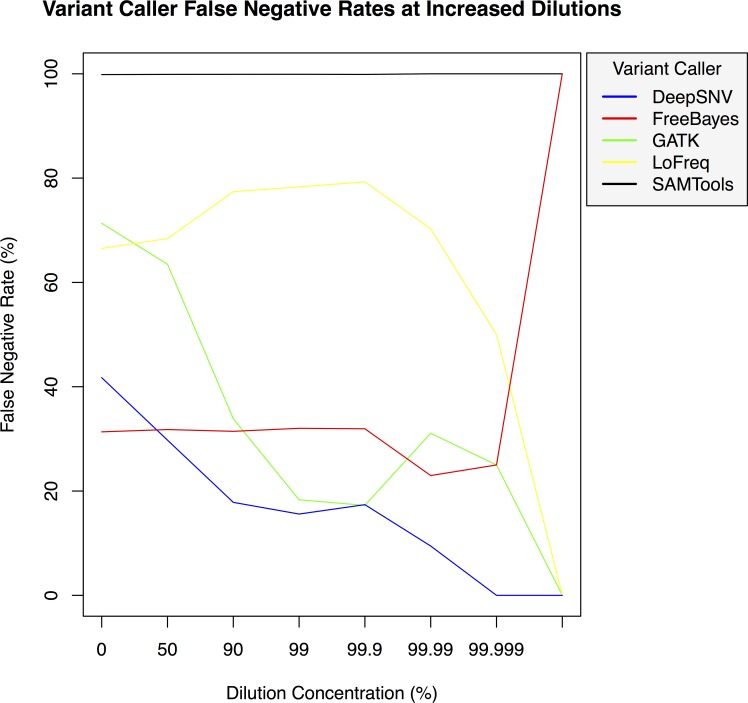
Background. Massively parallel sequencing technology is being used to sequence highly diverse populations of DNA such as that derived from heterogeneous cell mixtures containing both wild-type and disease-related states. At the core of such molecule tagging techniques is the tagging and identification of sequence reads derived from individual input DNA molecules, which must be first computationally disambiguated to generate read groups sharing common sequence tags, with each read group representing a single input DNA molecule. This disambiguation typically generates huge numbers of reads groups, each of which requires additional variant detection analysis steps to be run specific to each read group, thus representing a significant computational challenge. While sequencing technologies for producing these data are approaching maturity, the lack of available computational tools for analysing such heterogeneous sequence data represents an obstacle to the widespread adoption of this technology. Results. Using synthetic data we successfully detect unique variants at dilution levels of 1 in a 1,000,000 molecules, and find DeeepSNVMiner obtains significantly lower false positive and false negative rates compared to popular variant callers GATK, SAMTools, FreeBayes and LoFreq, particularly as the variant concentration levels decrease. In a dilution series with genomic DNA from two cells lines, we find DeepSNVMiner identifies a known somatic variant when present at concentrations of only 1 in 1,000 molecules in the input material, the lowest concentration amongst all variant callers tested. Conclusions. Here we present DeepSNVMiner; a tool to disambiguate tagged sequence groups and robustly identify sequence variants specific to subsets of starting DNA molecules that may indicate the presence of a disease. DeepSNVMiner is an automated workflow of custom sequence analysis utilities and open source tools able to differentiate somatic DNA variants from artefactual sequence variants that likely arose during DNA amplification. The workflow remains flexible such that it may be customised to variants of the data production protocol used, and supports reproducible analysis through detailed logging and reporting of results. DeepSNVMiner is available for academic non-commercial research purposes at https://github.com/mattmattmattmatt/DeepSNVMiner.
背景。大规模平行测序技术正被用于对高度多样化的DNA群体进行测序,比如来自包含野生型和疾病相关状态的异质细胞混合物的DNA。此类分子标记技术的核心是对源自单个输入DNA分子的序列读数进行标记和识别,这些读数必须首先通过计算进行解歧义处理,以生成共享共同序列标签的读数组,每个读数组代表一个单一的输入DNA分子。这种解歧义处理通常会产生大量的读数组,每个读数组都需要针对每个读数组运行特定的额外变异检测分析步骤,因此这是一项重大的计算挑战。虽然用于生成这些数据的测序技术已接近成熟,但缺乏用于分析此类异质序列数据的可用计算工具,这成为了该技术广泛应用的障碍。
结果。使用合成数据,我们成功地在百万分之一分子的稀释水平下检测到独特变异,并发现与流行的变异调用工具GATK、SAMTools、FreeBayes和LoFreq相比,DeeepSNVMiner获得的假阳性和假阴性率显著更低,尤其是随着变异浓度水平降低时。在使用来自两个细胞系的基因组DNA的稀释系列实验中,我们发现DeepSNVMiner在输入材料中仅以千分之一分子的浓度存在时就能识别出已知的体细胞变异,这是所有测试的变异调用工具中最低的浓度。
结论。在此我们展示了DeepSNVMiner;这是一种用于对标记的序列组进行解歧义处理并稳健识别特定于起始DNA分子子集的序列变异的工具,这些变异可能表明疾病的存在。DeepSNVMiner是一个由定制序列分析实用程序和开源工具组成的自动化工作流程,能够区分体细胞DNA变异与可能在DNA扩增过程中出现的人为序列变异。该工作流程保持灵活性,以便可以根据所使用的数据生产协议的变体进行定制,并通过详细的日志记录和结果报告支持可重复分析。DeepSNVMiner可用于学术非商业研究目的,网址为https://github.com/mattmattmattmatt/DeepSNVMiner。