Department of Computer Science, Stony Brook University, Stony Brook, NY, USA.
Bioinformatics. 2012 Aug 15;28(16):2097-105. doi: 10.1093/bioinformatics/bts330. Epub 2012 Jun 4.
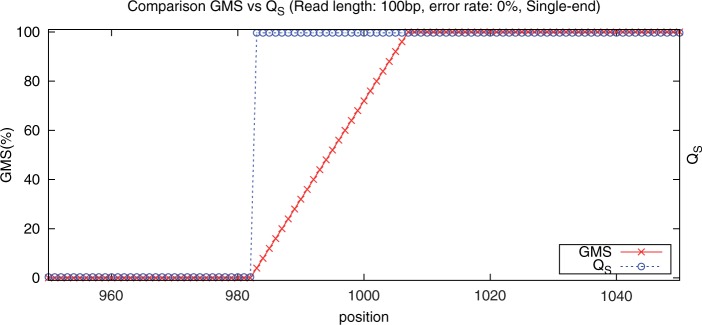
Genome resequencing and short read mapping are two of the primary tools of genomics and are used for many important applications. The current state-of-the-art in mapping uses the quality values and mapping quality scores to evaluate the reliability of the mapping. These attributes, however, are assigned to individual reads and do not directly measure the problematic repeats across the genome. Here, we present the Genome Mappability Score (GMS) as a novel measure of the complexity of resequencing a genome. The GMS is a weighted probability that any read could be unambiguously mapped to a given position and thus measures the overall composition of the genome itself.
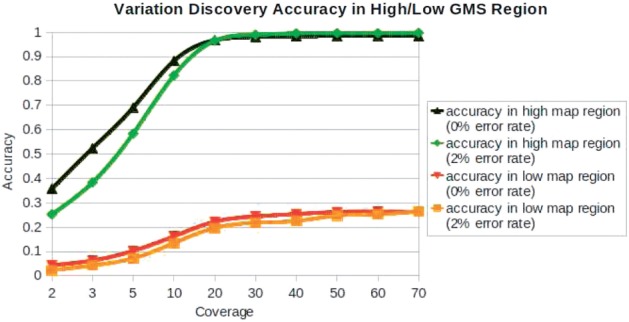
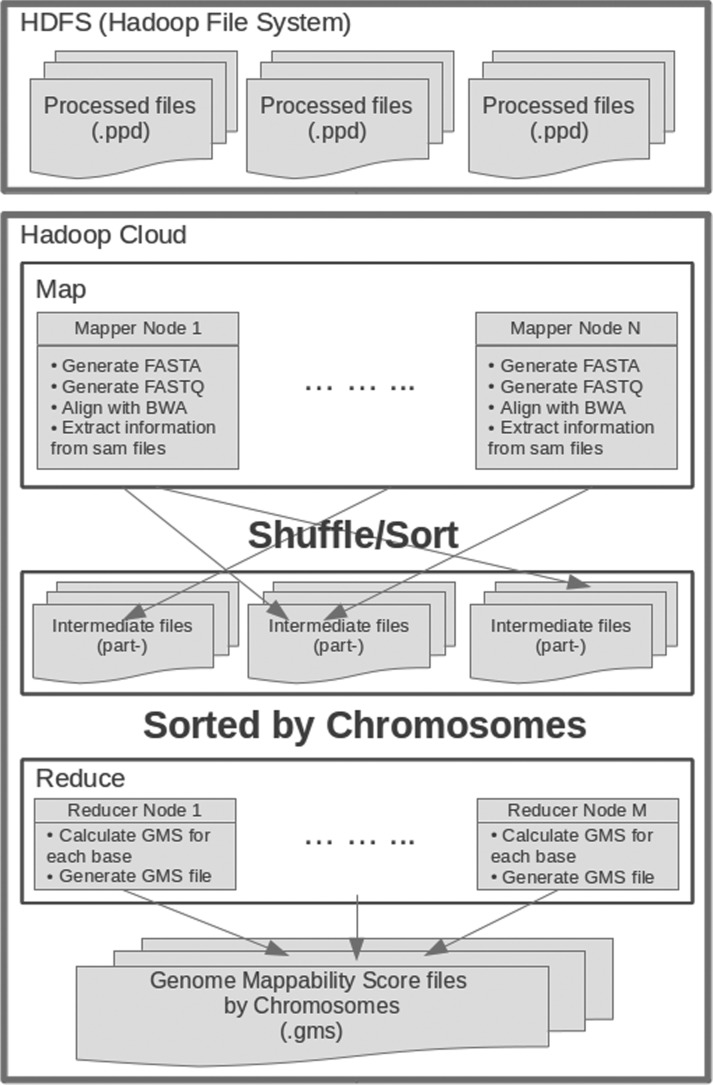
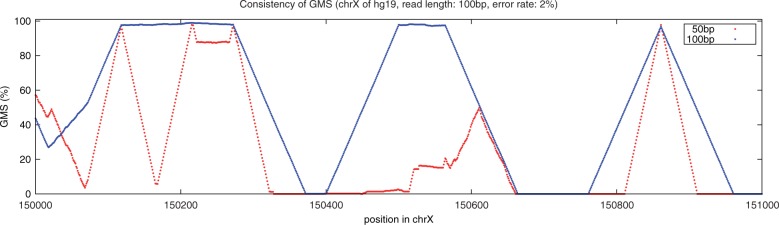
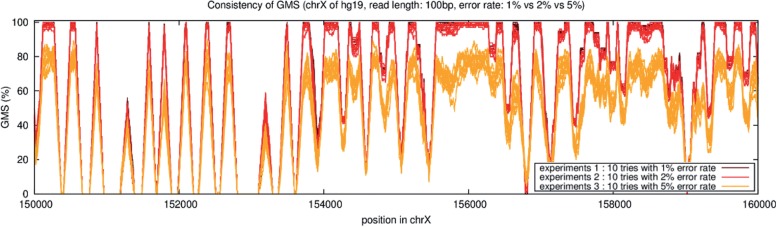
We have developed the Genome Mappability Analyzer to compute the GMS of every position in a genome. It leverages the parallelism of cloud computing to analyze large genomes, and enabled us to identify the 5-14% of the human, mouse, fly and yeast genomes that are difficult to analyze with short reads. We examined the accuracy of the widely used BWA/SAMtools polymorphism discovery pipeline in the context of the GMS, and found discovery errors are dominated by false negatives, especially in regions with poor GMS. These errors are fundamental to the mapping process and cannot be overcome by increasing coverage. As such, the GMS should be considered in every resequencing project to pinpoint the 'dark matter' of the genome, including of known clinically relevant variations in these regions.
The source code and profiles of several model organisms are available at http://gma-bio.sourceforge.net
基因组重测序和短读序列映射是基因组学的两种主要工具,可用于许多重要的应用。当前的映射技术使用质量值和映射质量分数来评估映射的可靠性。然而,这些属性是分配给单个读取的,并没有直接测量基因组中存在的问题重复。在这里,我们提出了基因组可映射性评分(GMS)作为重新测序基因组复杂性的新度量。GMS 是一个能够明确映射到给定位置的任何读取的加权概率,因此可以测量基因组本身的总体组成。
我们开发了基因组可映射性分析器来计算基因组中每个位置的 GMS。它利用云计算的并行性来分析大型基因组,并使我们能够识别人类、老鼠、苍蝇和酵母基因组中 5-14%的难以用短读序列进行分析的区域。我们在 GMS 背景下检查了广泛使用的 BWA/SAMtools 多态性发现管道的准确性,发现发现错误主要是假阴性,尤其是在 GMS 较差的区域。这些错误是映射过程的基础,不能通过增加覆盖度来克服。因此,在每个重测序项目中都应考虑 GMS,以查明基因组的“暗物质”,包括这些区域中已知的临床相关变异。
几种模型生物的源代码和图谱可在 http://gma-bio.sourceforge.net 上获得。