Garg Shilpa, Martin Marcel, Marschall Tobias
Center for Bioinformatics, Saarland University, Saarbrücken, Germany Max Planck Institute for Informatics, Saarbrücken, Germany Saarbrücken Graduate School of Computer Science, Saarland University, Saarbrücken, Germany.
Science for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, SE-17121 Solna, Sweden.
Bioinformatics. 2016 Jun 15;32(12):i234-i242. doi: 10.1093/bioinformatics/btw276.
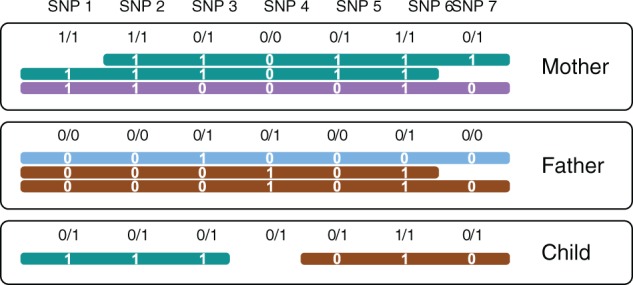
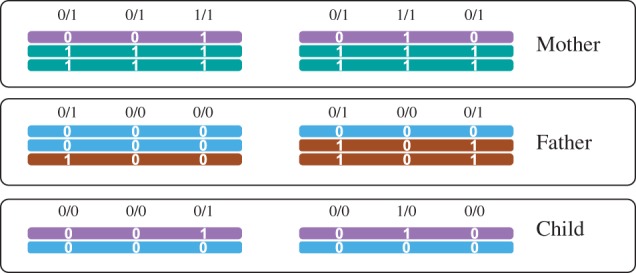
Read-based phasing deduces the haplotypes of an individual from sequencing reads that cover multiple variants, while genetic phasing takes only genotypes as input and applies the rules of Mendelian inheritance to infer haplotypes within a pedigree of individuals. Combining both into an approach that uses these two independent sources of information-reads and pedigree-has the potential to deliver results better than each individually.
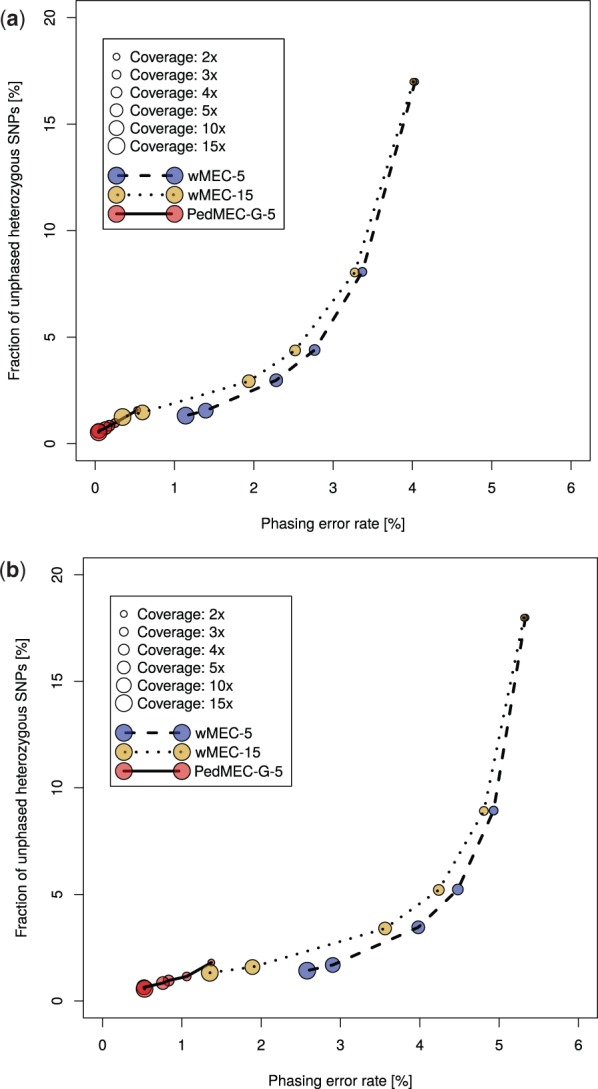
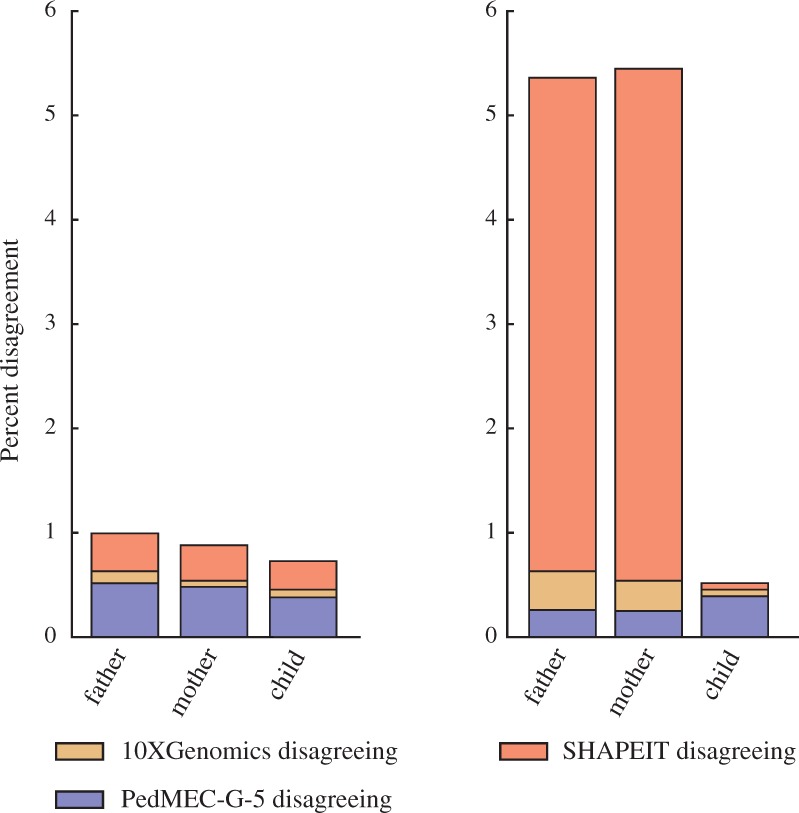
We provide a theoretical framework combining read-based phasing with genetic haplotyping, and describe a fixed-parameter algorithm and its implementation for finding an optimal solution. We show that leveraging reads of related individuals jointly in this way yields more phased variants and at a higher accuracy than when phased separately, both in simulated and real data. Coverages as low as 2× for each member of a trio yield haplotypes that are as accurate as when analyzed separately at 15× coverage per individual.
https://bitbucket.org/whatshap/whatshap
基于 reads 的定相分析从覆盖多个变异的测序 reads 中推断个体的单倍型,而基因定相分析仅将基因型作为输入,并应用孟德尔遗传规则在个体家系中推断单倍型。将这两种方法结合成一种使用这两种独立信息源(reads 和家系)的方法,有可能产生比单独使用每种方法更好的结果。
我们提供了一个将基于 reads 的定相分析与基因单倍型分析相结合的理论框架,并描述了一种用于找到最优解的固定参数算法及其实现。我们表明,在模拟数据和真实数据中,以这种方式联合利用相关个体的 reads 比单独定相时能产生更多的定相变异,且准确性更高。三人组中每个成员低至 2× 的覆盖度就能产生与个体单独以 15× 覆盖度分析时一样准确的单倍型。
https://bitbucket.org/whatshap/whatshap