Department of Biology, Brigham Young University, Provo, UT, 84602, USA.
BMC Bioinformatics. 2021 Nov 22;22(1):559. doi: 10.1186/s12859-021-04470-4.
When analyzing DNA sequence data of an individual, knowing which nucleotide was inherited from each parent can be beneficial when trying to identify certain types of DNA variants. Mendelian inheritance logic can be used to accurately phase (haplotype) the majority (67-83%) of an individual's heterozygous nucleotide positions when genotypes are available for both parents (trio). However, when all members of a trio are heterozygous at a position, Mendelian inheritance logic cannot be used to phase. For such positions, a computational phasing algorithm can be used. Existing phasing algorithms use a haplotype reference panel, sequencing reads, and/or parental genotypes to phase an individual; however, they are limited in that they can only phase certain types of variants, require a specific genotype build, require large amounts of storage capacity, and/or require long run times. We created trioPhaser to address these challenges.
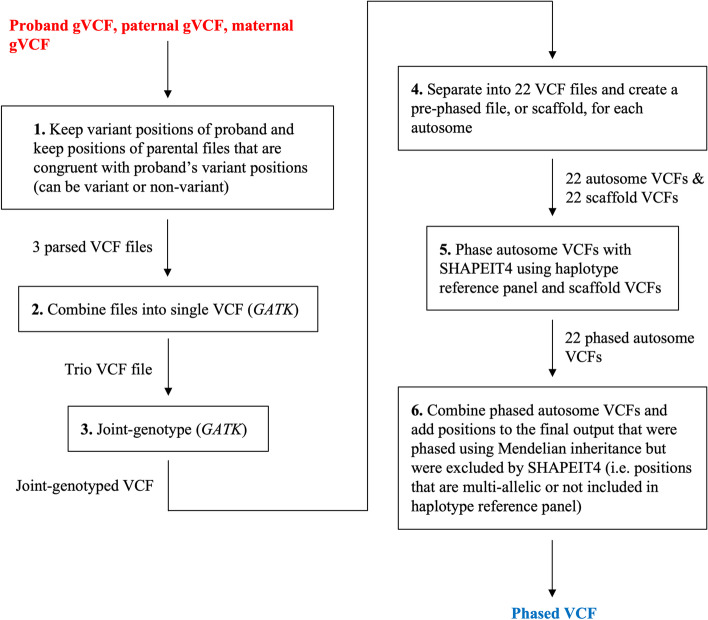
trioPhaser uses gVCF files from an individual and their parents as initial input, and then outputs a phased VCF file. Input trio data are first phased using Mendelian inheritance logic. Then, the positions that cannot be phased using inheritance information alone are phased by the SHAPEIT4 phasing algorithm. Using whole-genome sequencing data of 52 trios, we show that trioPhaser, on average, increases the total number of phased positions by 21.0% and 10.5%, respectively, when compared to the number of positions that SHAPEIT4 or Mendelian inheritance logic can phase when either is used alone. In addition, we show that the accuracy of the phased calls output by trioPhaser are similar to linked-read and read-backed phasing.
trioPhaser is a containerized software tool that uses both Mendelian inheritance logic and SHAPEIT4 to phase trios when gVCF files are available. By implementing both phasing methods, more variant positions are phased compared to what either method is able to phase alone.
当分析个体的 DNA 序列数据时,了解每个亲本遗传的核苷酸可以帮助识别某些类型的 DNA 变体。当父母双方(三亲)的基因型可用时,可以使用孟德尔遗传逻辑准确地对个体的大多数(67-83%)杂合核苷酸位置进行相位(单倍型)。然而,当三亲的所有成员在一个位置都是杂合子时,就不能使用孟德尔遗传逻辑进行相位。对于这种情况,可以使用计算相位算法。现有的相位算法使用单倍型参考面板、测序reads 和/或亲本基因型来对个体进行相位;然而,它们受到限制,因为它们只能相位特定类型的变体,需要特定的基因型构建,需要大量的存储容量,并且/或者需要长时间运行。我们创建了 trioPhaser 来解决这些挑战。
trioPhaser 使用个体及其父母的 gVCF 文件作为初始输入,然后输出一个相位 VCF 文件。输入的三亲数据首先使用孟德尔遗传逻辑进行相位。然后,无法仅使用遗传信息进行相位的位置由 SHAPEIT4 相位算法进行相位。使用 52 个三亲的全基因组测序数据,我们表明,与仅使用 SHAPEIT4 或孟德尔遗传逻辑时可以相位的位置数量相比,trioPhaser 平均分别将相位位置的总数增加了 21.0%和 10.5%。此外,我们表明 trioPhaser 输出的相位调用的准确性与链接读取和读取回推相位相似。
trioPhaser 是一个容器化软件工具,当有 gVCF 文件时,它使用孟德尔遗传逻辑和 SHAPEIT4 对三亲进行相位。通过同时实现这两种相位方法,与每种方法单独相位相比,更多的变体位置被相位。