Pearson Richard D, Amato Roberto, Auburn Sarah, Miotto Olivo, Almagro-Garcia Jacob, Amaratunga Chanaki, Suon Seila, Mao Sivanna, Noviyanti Rintis, Trimarsanto Hidayat, Marfurt Jutta, Anstey Nicholas M, William Timothy, Boni Maciej F, Dolecek Christiane, Hien Tinh Tran, White Nicholas J, Michon Pascal, Siba Peter, Tavul Livingstone, Harrison Gabrielle, Barry Alyssa, Mueller Ivo, Ferreira Marcelo U, Karunaweera Nadira, Randrianarivelojosia Milijaona, Gao Qi, Hubbart Christina, Hart Lee, Jeffery Ben, Drury Eleanor, Mead Daniel, Kekre Mihir, Campino Susana, Manske Magnus, Cornelius Victoria J, MacInnis Bronwyn, Rockett Kirk A, Miles Alistair, Rayner Julian C, Fairhurst Rick M, Nosten Francois, Price Ric N, Kwiatkowski Dominic P
Wellcome Trust Sanger Institute, Hinxton, Cambridge CB10 1SA, UK.
MRC Centre for Genomics and Global Health, Wellcome Trust Centre for Human Genetics, Oxford OX3 7BN, UK.
Nat Genet. 2016 Aug;48(8):959-964. doi: 10.1038/ng.3599. Epub 2016 Jun 27.
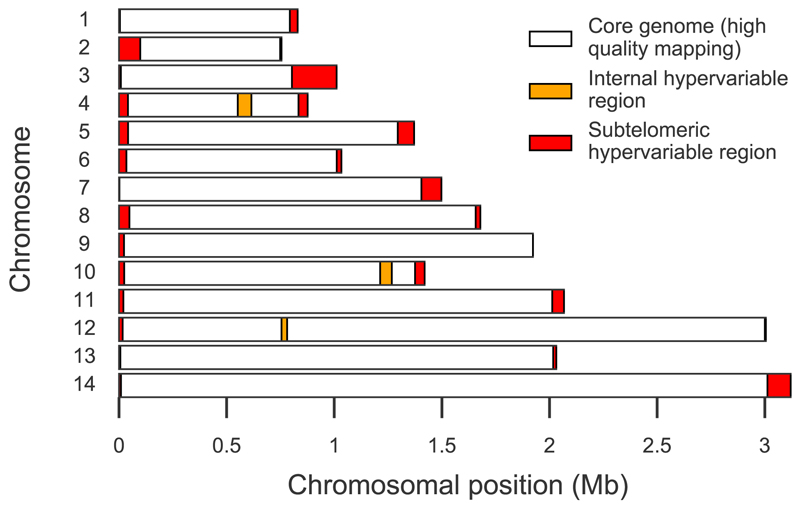
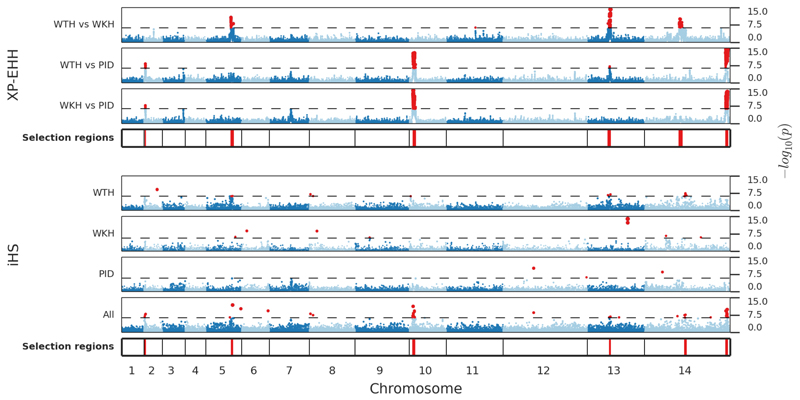
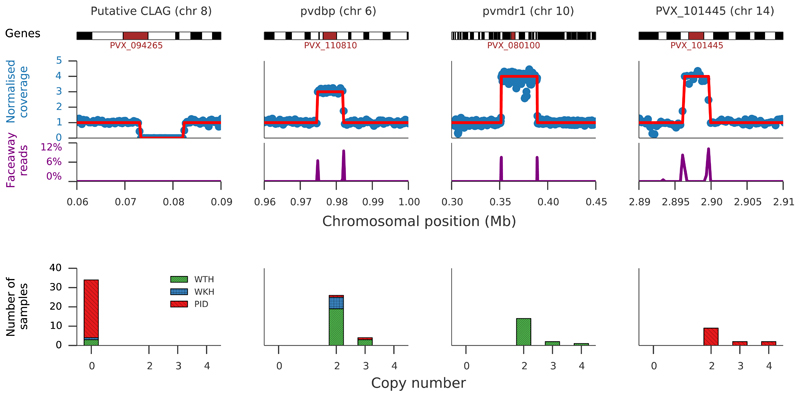
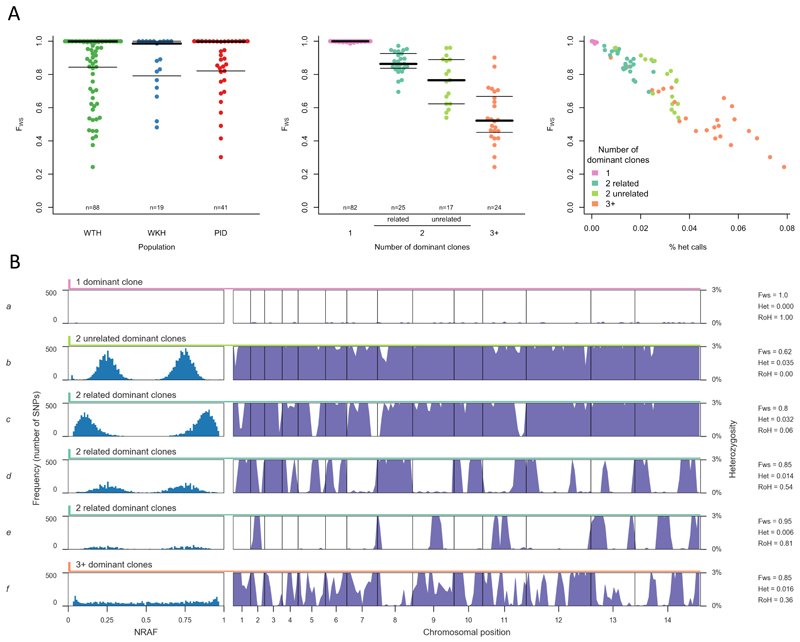
The widespread distribution and relapsing nature of Plasmodium vivax infection present major challenges for the elimination of malaria. To characterize the genetic diversity of this parasite in individual infections and across the population, we performed deep genome sequencing of >200 clinical samples collected across the Asia-Pacific region and analyzed data on >300,000 SNPs and nine regions of the genome with large copy number variations. Individual infections showed complex patterns of genetic structure, with variation not only in the number of dominant clones but also in their level of relatedness and inbreeding. At the population level, we observed strong signals of recent evolutionary selection both in known drug resistance genes and at new loci, and these varied markedly between geographical locations. These findings demonstrate a dynamic landscape of local evolutionary adaptation in the parasite population and provide a foundation for genomic surveillance to guide effective strategies for control and elimination of P. vivax.
间日疟原虫感染的广泛分布和复发性给疟疾消除带来了重大挑战。为了表征这种寄生虫在个体感染及整个人口中的遗传多样性,我们对亚太地区收集的200多个临床样本进行了深度基因组测序,并分析了30多万个单核苷酸多态性(SNP)以及基因组中9个具有大量拷贝数变异的区域的数据。个体感染呈现出复杂的遗传结构模式,不仅优势克隆的数量存在差异,而且它们的亲缘关系和近亲繁殖程度也有所不同。在种群水平上,我们在已知的耐药基因和新位点均观察到近期进化选择的强烈信号,并且这些信号在地理位置之间存在显著差异。这些发现揭示了寄生虫种群中局部进化适应的动态图景,并为基因组监测提供了基础,以指导控制和消除间日疟原虫的有效策略。