Division of Infectious Diseases, Department of Medicine, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA, 92093, USA.
U.S. Naval Medical Research No. 6, Venezuela Ave, Block 36, Bellavista, Callao, Peru.
Genome Med. 2018 Jul 4;10(1):52. doi: 10.1186/s13073-018-0563-0.
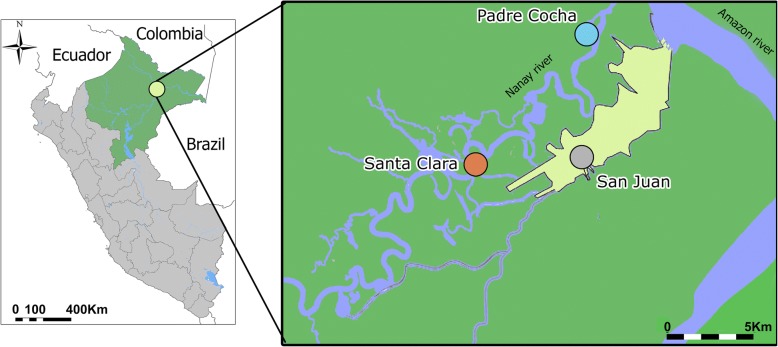
Plasmodium vivax poses a significant challenge to malaria elimination due to its ability to cause relapsed infections from reactivation of dormant liver parasites called hypnozoites. We analyzed 69 P. vivax whole genome sequences obtained from subjects residing in three different villages along the Peruvian Amazon. This included 23 paired P. vivax samples from subjects who experienced recurrent P. vivax parasitemia following observed treatment with chloroquine and primaquine.
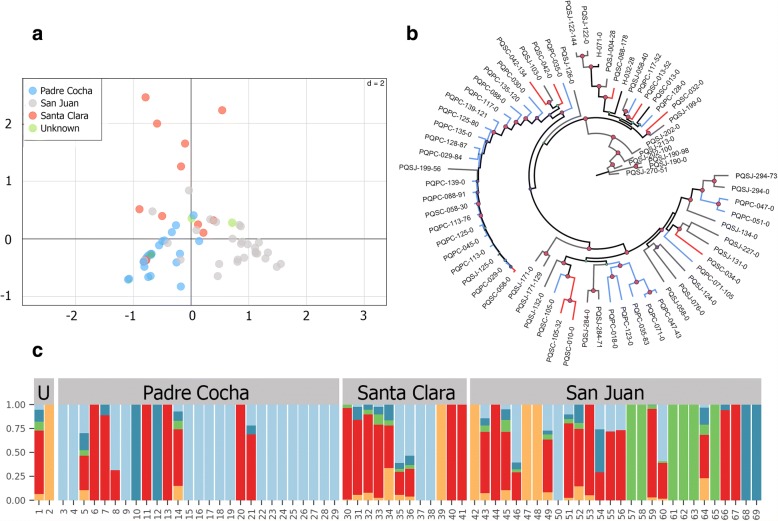
Genomic DNA was extracted from whole blood samples collected from subjects. P. vivax DNA was enriched using selective whole genome amplification and whole genome sequencing. We used single nucleotide polymorphisms (SNPs) from the core P. vivax genome to determine characteristics of the parasite population using discriminant analysis of principal components, maximum likelihood estimation of individual ancestries, and phylogenetic analysis. We estimated the relatedness of the paired samples by calculating the number of segregating sites and using a hidden Markov model approach to estimate identity by descent.
We present a comprehensive dataset of population genetics of Plasmodium vivax in the Peruvian Amazonian. We define the parasite population structure in this region and demonstrate a novel method for distinguishing homologous relapses from reinfections or heterologous relapses with improved accuracy. The parasite population in this area was quite diverse with an estimated five subpopulations and evidence of a highly heterogeneous ancestry of some of the isolates, similar to previous analyses of P. vivax in this region. Pairwise comparison of recurrent infections determined that there were 12 homologous relapses and 3 likely heterologous relapses with highly related parasites. To the best of our knowledge, this is the first large-scale study to evaluate recurrent P. vivax infections using whole genome sequencing.
Whole genome sequencing is a high-resolution tool that can identify P. vivax homologous relapses with increased sensitivity, while also providing data about drug resistance and parasite population genetics. This information is important for evaluating the efficacy of known and novel antirelapse medications in endemic areas and thus advancing the campaign to eliminate malaria.
间日疟原虫(Plasmodium vivax)因其休眠肝脏寄生虫(称为休眠子)重新激活而导致复发感染的能力,给疟疾消除工作带来了巨大挑战。我们分析了来自秘鲁亚马逊地区三个不同村庄的 69 株间日疟原虫全基因组序列,这些序列来自于接受氯喹和伯氨喹治疗后出现复发性间日疟原虫血症的 23 对个体。
从研究对象采集的全血样本中提取基因组 DNA。使用选择性全基因组扩增和全基因组测序来富集间日疟原虫 DNA。我们使用核心间日疟原虫基因组中的单核苷酸多态性(SNP),通过主成分判别分析、个体起源的最大似然估计和系统发育分析来确定寄生虫群体的特征。我们通过计算分离位点的数量并使用隐马尔可夫模型方法来估计血缘关系,来估算配对样本的相关性。
我们提供了一个关于秘鲁亚马逊地区间日疟原虫种群遗传学的综合数据集。我们定义了该地区的寄生虫种群结构,并展示了一种从同源复发中区分同源复发、再感染或异源复发的新方法,该方法具有更高的准确性。该地区的寄生虫群体非常多样化,估计有 5 个亚群,并且一些分离株的起源具有高度异质性,这与该地区以前对间日疟原虫的分析相似。对复发性感染的成对比较表明,有 12 次同源复发和 3 次可能的异源复发,与高度相关的寄生虫有关。据我们所知,这是首次使用全基因组测序评估复发性间日疟原虫感染的大规模研究。
全基因组测序是一种高分辨率工具,可以提高识别间日疟原虫同源复发的敏感性,同时还提供有关药物耐药性和寄生虫种群遗传学的信息。这些信息对于评估在流行地区已知和新型抗复发药物的疗效以及推进消除疟疾的运动非常重要。