Guangdong-Hongkong-Macau Institute of CNS Regeneration, Jinan University, Guangzhou 510632, China.
Ministry of Education Joint International Research Laboratory of CNS Regeneration, Jinan University, Guangzhou 510632, China.
Nat Commun. 2016 Jun 30;7:12065. doi: 10.1038/ncomms12065.
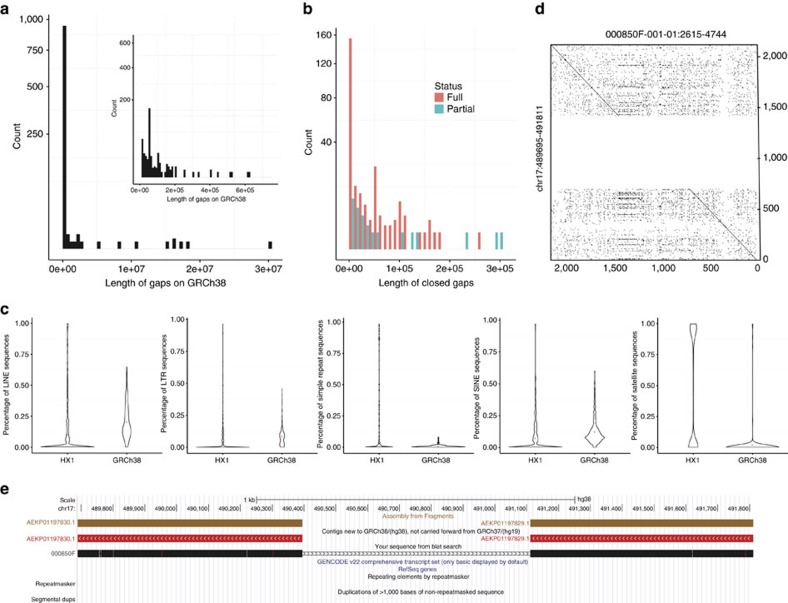
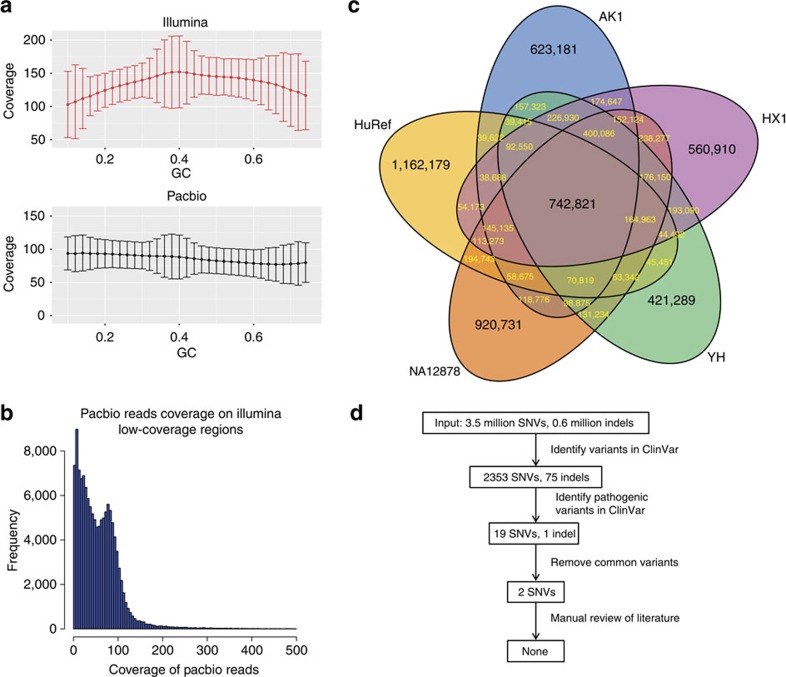
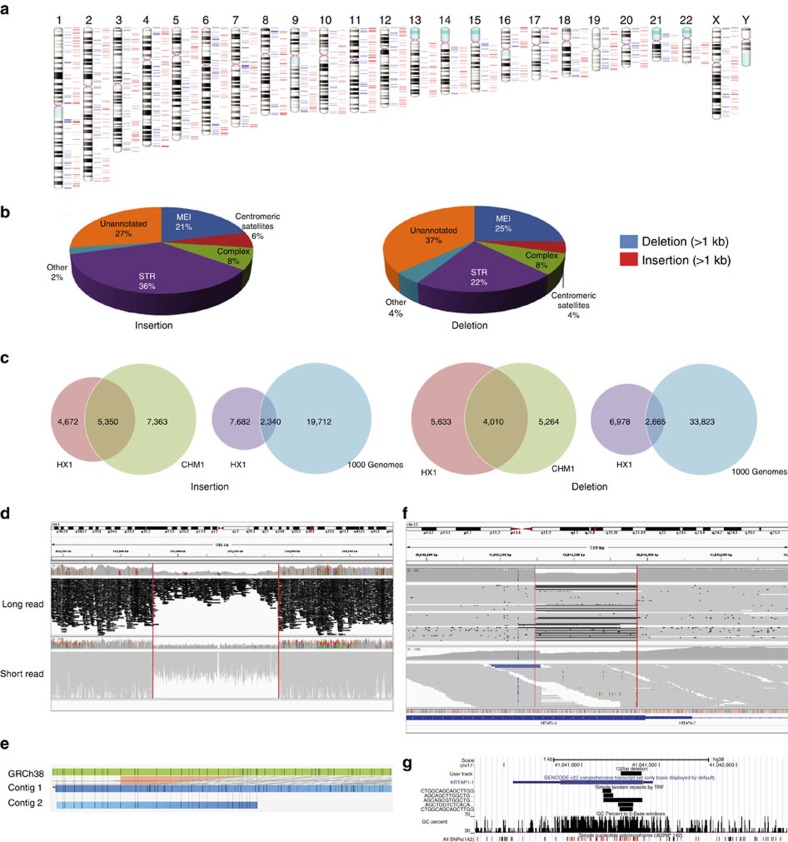
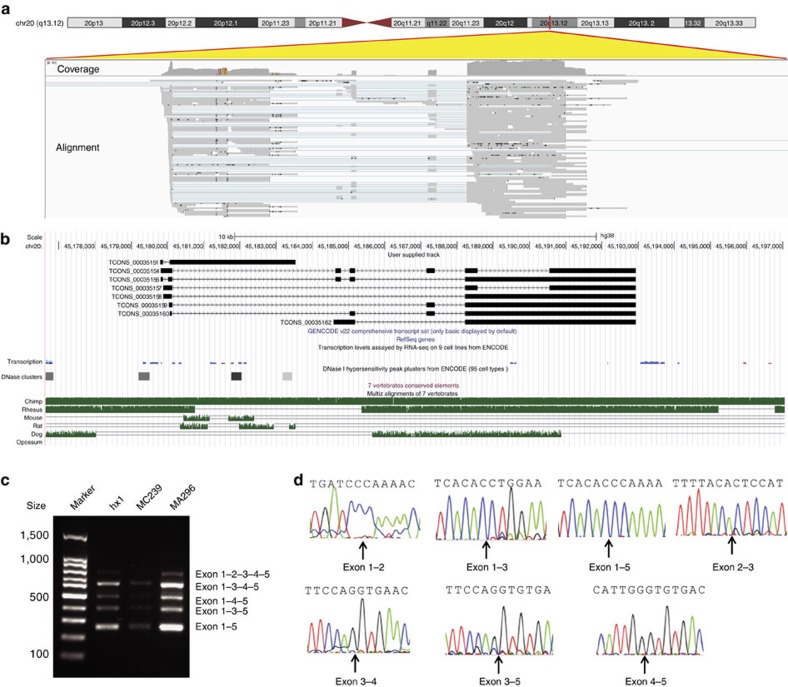
Short-read sequencing has enabled the de novo assembly of several individual human genomes, but with inherent limitations in characterizing repeat elements. Here we sequence a Chinese individual HX1 by single-molecule real-time (SMRT) long-read sequencing, construct a physical map by NanoChannel arrays and generate a de novo assembly of 2.93 Gb (contig N50: 8.3 Mb, scaffold N50: 22.0 Mb, including 39.3 Mb N-bases), together with 206 Mb of alternative haplotypes. The assembly fully or partially fills 274 (28.4%) N-gaps in the reference genome GRCh38. Comparison to GRCh38 reveals 12.8 Mb of HX1-specific sequences, including 4.1 Mb that are not present in previously reported Asian genomes. Furthermore, long-read sequencing of the transcriptome reveals novel spliced genes that are not annotated in GENCODE and are missed by short-read RNA-Seq. Our results imply that improved characterization of genome functional variation may require the use of a range of genomic technologies on diverse human populations.
短读测序技术已经能够从头组装多个个体的人类基因组,但在描述重复元件方面存在固有局限性。在这里,我们通过单分子实时(SMRT)长读测序对中国个体 HX1 进行测序,通过纳米通道阵列构建物理图谱,并生成 2.93Gb 的从头组装(contig N50:8.3Mb,scaffold N50:22.0Mb,包括 39.3Mb N-碱基),以及 206Mb 的替代单倍型。该组装完全或部分填补了参考基因组 GRCh38 中的 274 个 N 缺口(占 28.4%)。与 GRCh38 的比较显示,HX1 有 12.8Mb 的特异性序列,其中 4.1Mb 不存在于之前报道的亚洲基因组中。此外,转录组的长读测序揭示了新的剪接基因,这些基因在 GENCODE 中没有注释,也被短读 RNA-Seq 遗漏。我们的结果表明,要更好地描述基因组功能变异,可能需要在不同的人类群体中使用一系列基因组技术。