Rodrigo María Belén, Mojsiejczuk Laura Noelia, Torres Carolina, Sevic Ina, González López Ledesma María Mora, Perez Paula Soledad, Bouzas María Belén, Galdame Omar, Marciano Sebastián, Fainboim Hugo, Flichman Diego Martín, Campos Rodolfo Héctor
Cátedra de Virología, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Argentina, Ciudad Autónoma de Buenos Aires, Argentina.
Unidad de Virología, Hospital de Enfermedades Infecciosas ''F. Muñiz", Ciudad Autónoma de Buenos Aires, Argentina.
PLoS One. 2016 Jul 19;11(7):e0159509. doi: 10.1371/journal.pone.0159509. eCollection 2016.
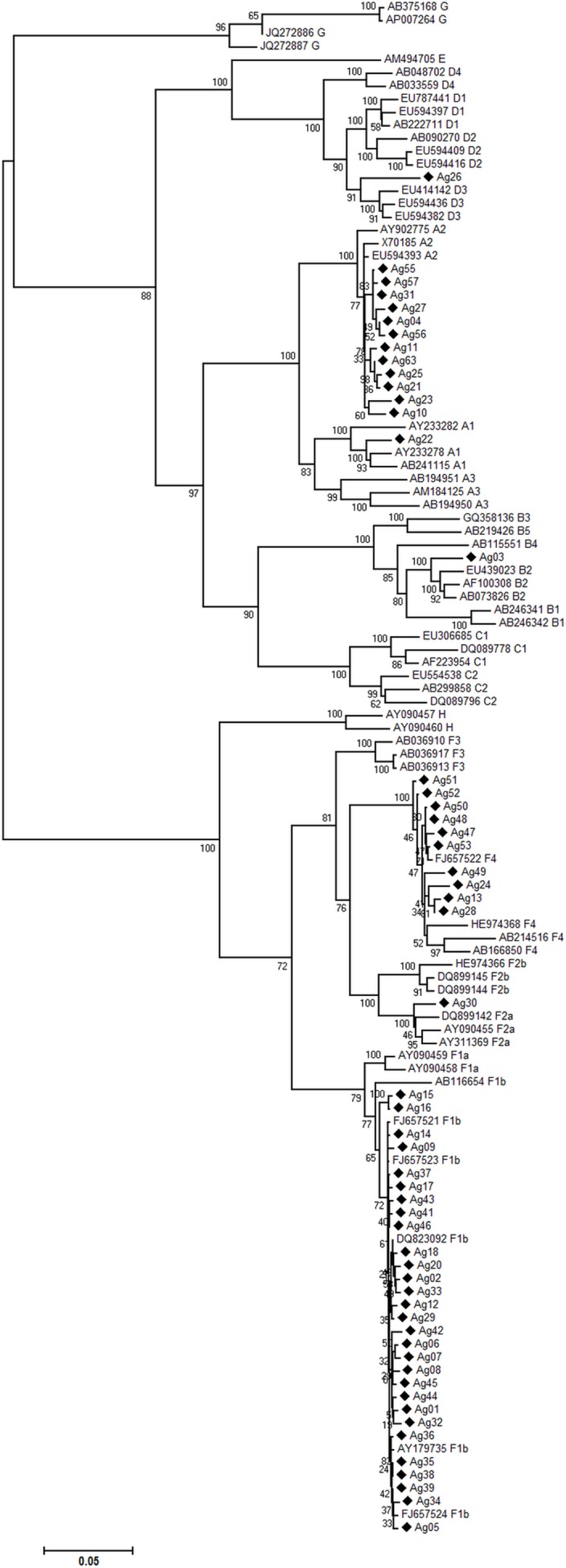
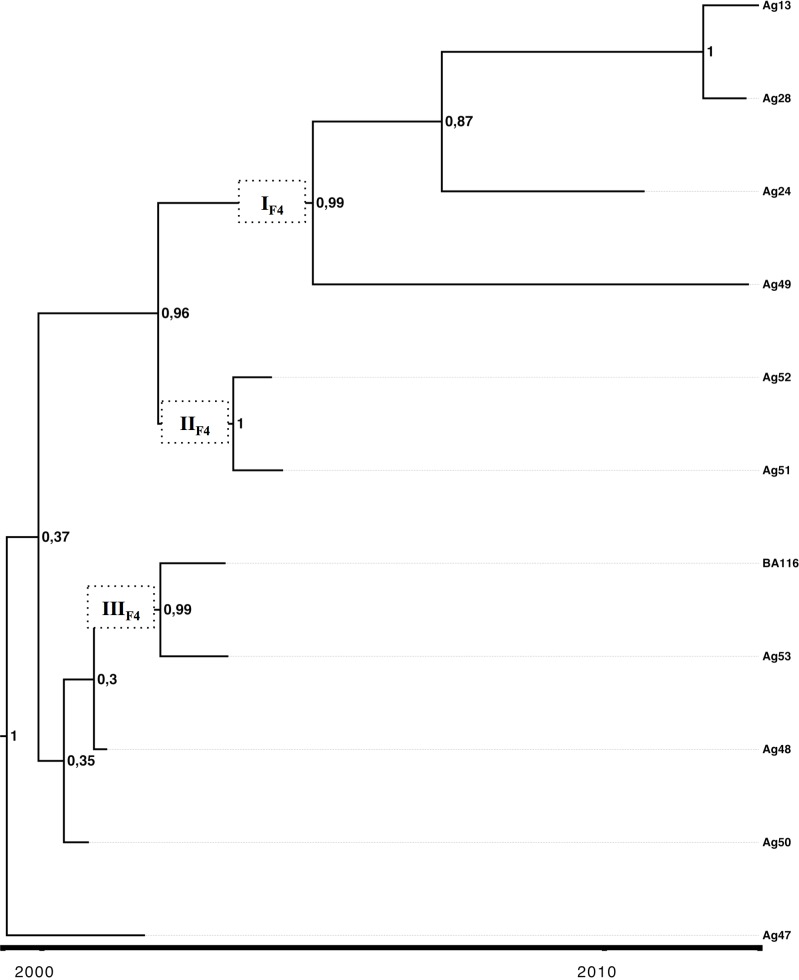
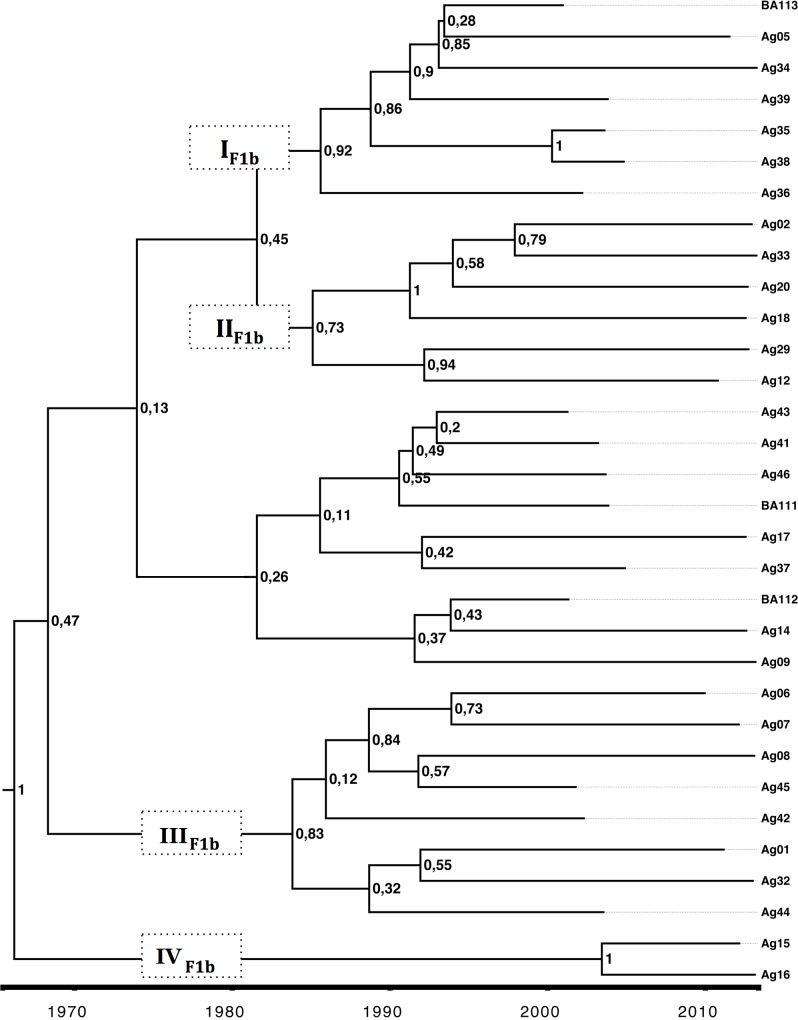
Hepatitis B virus (HBV) is a globally distributed human pathogen that leads to both self-limited and chronic infections. At least eight genotypes (A-H) with distinct geographical allocations and phylodynamic behaviors have been described. They differ substantially in many virological and probably some clinical parameters. The aim of this study was to analyze full-length HBV genome sequences from individuals with symptomatic acute HBV infections using phylogenetic and coalescent methods. The phylogenetic analysis resulted in the following subgenotype distribution: F1b (52.7%), A2 (18.2%), F4 (18.2%) and A1, B2, D3 and F2a 1.8% each. These results contrast with those previously reported from chronic infections, where subgenotypes F1b, F4, A2 and genotype D were evenly distributed. This differential distribution might be related to recent internal migrations and/or intrinsic biological features of each viral genotype that could impact on the probability of transmission. The coalescence analysis showed that after a diversification process started in the 80s, the current sequences of subgenotype F1b were grouped in at least four highly supported lineages, whereas subgenotype F4 revealed a more limited diversification pattern with most lineages without offspring in the present. In addition, the genetic characterization of the studied sequences showed that only two of them presented mutations of clinical relevance at S codifyng region and none at the polymerase catalytic domains. Finally, since the acute infections could be an expression of the genotypes currently being transmitted to new hosts, the predominance of subgenotype F1b might have epidemiological, as well as, clinical relevance due to its potential adverse disease outcome among the chronic cases.
乙型肝炎病毒(HBV)是一种全球分布的人类病原体,可导致自限性感染和慢性感染。目前已描述了至少八种基因型(A - H),它们具有不同的地理分布和系统发育动力学行为。它们在许多病毒学参数以及可能的一些临床参数上存在很大差异。本研究的目的是使用系统发育和溯祖方法分析有症状急性HBV感染个体的全长HBV基因组序列。系统发育分析得出以下亚基因型分布:F1b(52.7%)、A2(18.2%)、F4(18.2%)以及A1、B2、D3和F2a各占1.8%。这些结果与先前报道的慢性感染结果形成对比,在慢性感染中,亚基因型F1b、F4、A2和基因型D分布均匀。这种差异分布可能与近期的国内迁移和/或每种病毒基因型的内在生物学特征有关,这些特征可能会影响传播概率。溯祖分析表明,在80年代开始的多样化过程之后,亚基因型F1b的当前序列至少分为四个得到高度支持的谱系,而亚基因型F4显示出更有限的多样化模式,目前大多数谱系没有后代。此外,对所研究序列的基因特征分析表明,其中只有两个在S编码区出现了具有临床相关性的突变,而在聚合酶催化结构域没有出现此类突变。最后,由于急性感染可能是当前正在传播给新宿主的基因型的一种表现形式,亚基因型F1b的优势可能具有流行病学以及临床相关性,因为它在慢性病例中可能导致不良疾病结局。