Choi Wonyoung, Lee Jungwoo, Lee Jin-Young, Lee Sun-Min, Kim Da-Won, Kim Young-Joon
Department of Integrated OMICS for Biomedical Science, Graduate School, Yonsei University, Seoul 03722, Korea.
Department of Biochemistry, College of Life Science & Biotechnology, Yonsei University, Seoul 03722, Korea.
Genomics Inform. 2016 Jun;14(2):46-52. doi: 10.5808/GI.2016.14.2.46. Epub 2016 Jun 30.
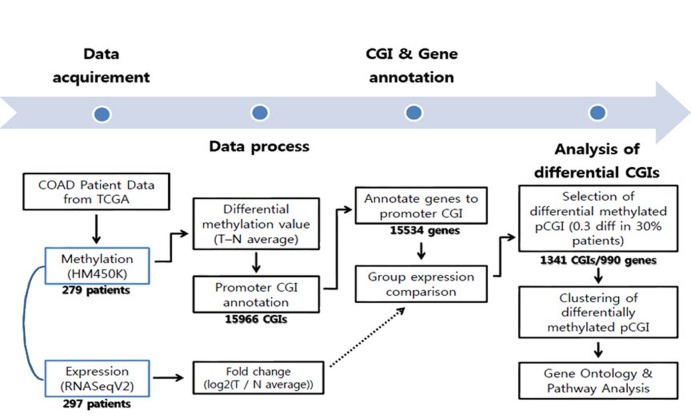
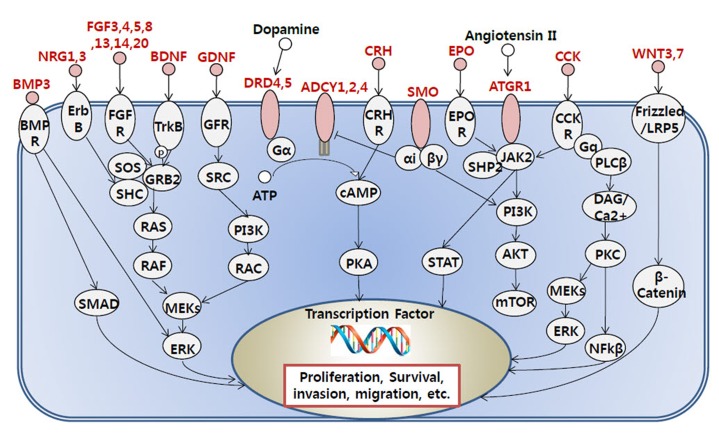
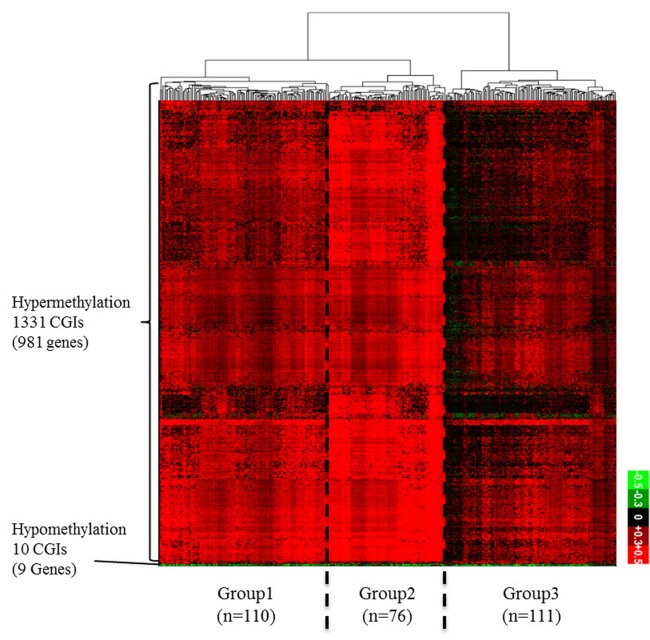
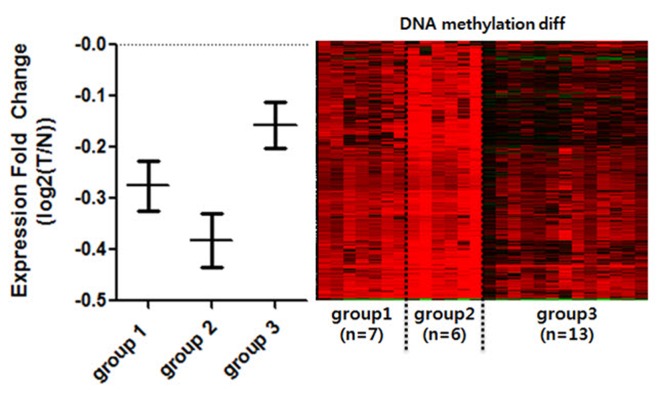
Diverse somatic mutations have been reported to serve as cancer drivers. Recently, it has also been reported that epigenetic regulation is closely related to cancer development. However, the effect of epigenetic changes on cancer is still elusive. In this study, we analyzed DNA methylation data on colon cancer taken from The Caner Genome Atlas. We found that several promoters were significantly hypermethylated in colon cancer patients. Through clustering analysis of differentially methylated DNA regions, we were able to define subgroups of patients and observed clinical features associated with each subgroup. In addition, we analyzed the functional ontology of aberrantly methylated genes and identified the G-protein-coupled receptor signaling pathway as one of the major pathways affected epigenetically. In conclusion, our analysis shows the possibility of characterizing the clinical features of colon cancer subgroups based on DNA methylation patterns and provides lists of important genes and pathways possibly involved in colon cancer development.
据报道,多种体细胞突变可作为癌症驱动因素。最近,也有报道称表观遗传调控与癌症发展密切相关。然而,表观遗传变化对癌症的影响仍不清楚。在本研究中,我们分析了来自癌症基因组图谱(The Cancer Genome Atlas)的结肠癌DNA甲基化数据。我们发现,在结肠癌患者中,有几个启动子显著高甲基化。通过对差异甲基化DNA区域进行聚类分析,我们能够定义患者亚组,并观察到与每个亚组相关的临床特征。此外,我们分析了异常甲基化基因的功能本体,确定G蛋白偶联受体信号通路是受表观遗传影响的主要通路之一。总之,我们的分析表明,基于DNA甲基化模式来表征结肠癌亚组临床特征是有可能的,并提供了可能参与结肠癌发展的重要基因和通路清单。