Gao Chuan, McDowell Ian C, Zhao Shiwen, Brown Christopher D, Engelhardt Barbara E
Department of Statistical Science, Duke University, Durham, North Carolina, United States of America.
Program in Computational Biology and Bioinformatics, Duke University, Durham, North Carolina, United States of America.
PLoS Comput Biol. 2016 Jul 28;12(7):e1004791. doi: 10.1371/journal.pcbi.1004791. eCollection 2016 Jul.
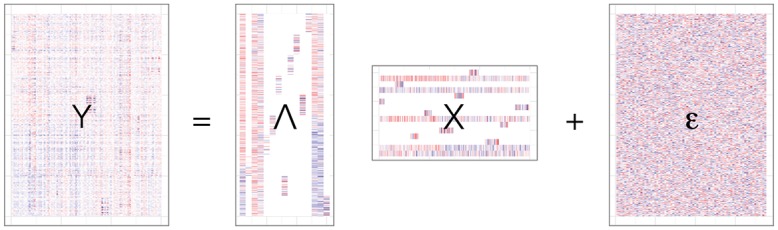
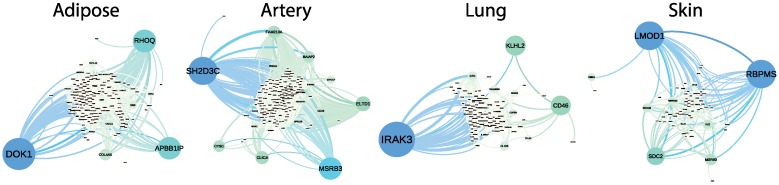
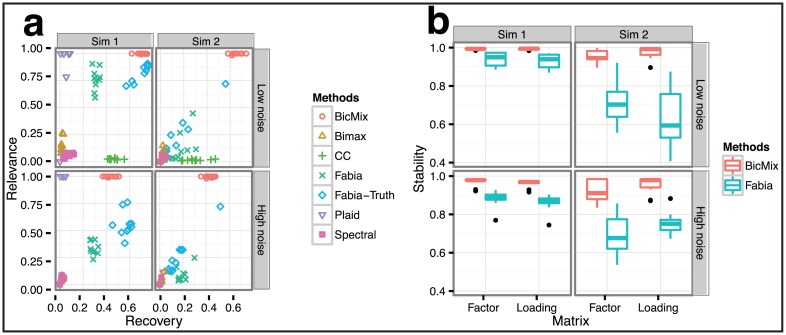
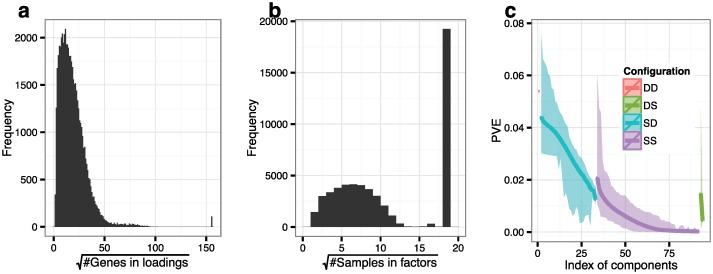
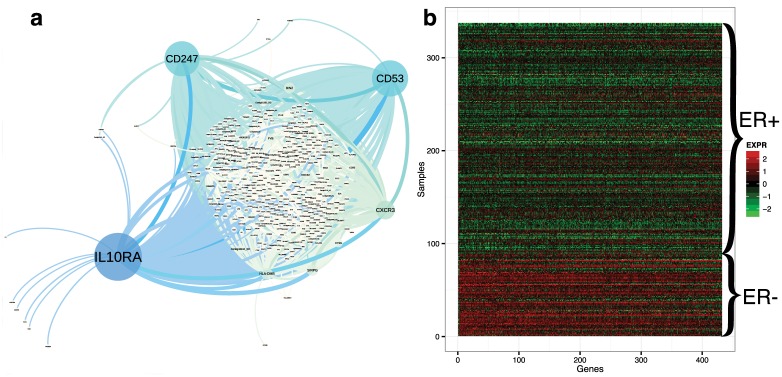
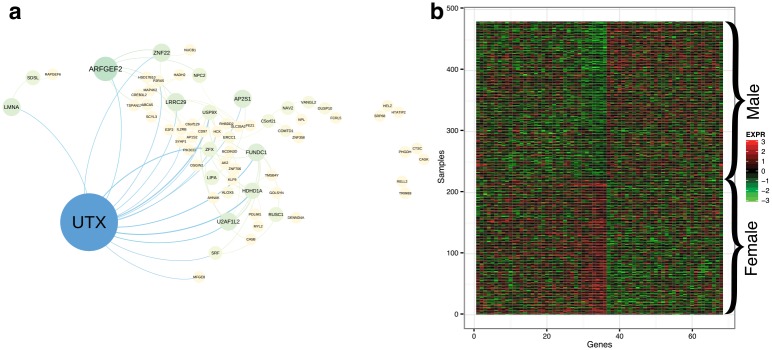
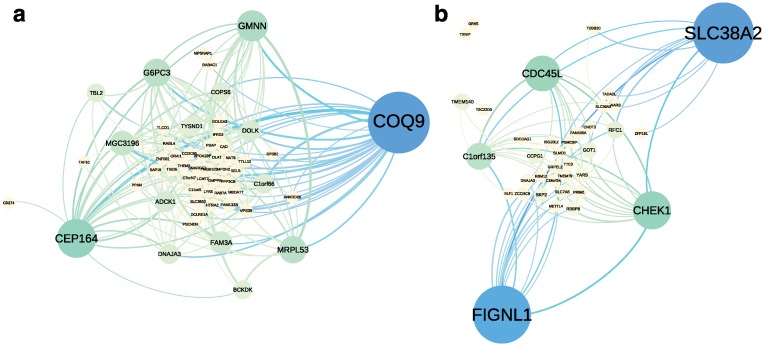
Identifying latent structure in high-dimensional genomic data is essential for exploring biological processes. Here, we consider recovering gene co-expression networks from gene expression data, where each network encodes relationships between genes that are co-regulated by shared biological mechanisms. To do this, we develop a Bayesian statistical model for biclustering to infer subsets of co-regulated genes that covary in all of the samples or in only a subset of the samples. Our biclustering method, BicMix, allows overcomplete representations of the data, computational tractability, and joint modeling of unknown confounders and biological signals. Compared with related biclustering methods, BicMix recovers latent structure with higher precision across diverse simulation scenarios as compared to state-of-the-art biclustering methods. Further, we develop a principled method to recover context specific gene co-expression networks from the estimated sparse biclustering matrices. We apply BicMix to breast cancer gene expression data and to gene expression data from a cardiovascular study cohort, and we recover gene co-expression networks that are differential across ER+ and ER- samples and across male and female samples. We apply BicMix to the Genotype-Tissue Expression (GTEx) pilot data, and we find tissue specific gene networks. We validate these findings by using our tissue specific networks to identify trans-eQTLs specific to one of four primary tissues.
识别高维基因组数据中的潜在结构对于探索生物过程至关重要。在此,我们考虑从基因表达数据中恢复基因共表达网络,其中每个网络编码由共享生物机制共同调控的基因之间的关系。为此,我们开发了一种用于双聚类的贝叶斯统计模型,以推断在所有样本或仅在样本的一个子集中协变的共同调控基因的子集。我们的双聚类方法BicMix允许对数据进行过完备表示、计算易处理性以及对未知混杂因素和生物信号进行联合建模。与相关双聚类方法相比,在各种模拟场景下,BicMix与最先进的双聚类方法相比,能以更高的精度恢复潜在结构。此外,我们开发了一种有原则的方法,从估计的稀疏双聚类矩阵中恢复特定上下文的基因共表达网络。我们将BicMix应用于乳腺癌基因表达数据和心血管研究队列的基因表达数据,并恢复了在ER +和ER -样本以及男性和女性样本之间存在差异的基因共表达网络。我们将BicMix应用于基因型 - 组织表达(GTEx)试点数据,并发现了组织特异性基因网络。我们通过使用我们的组织特异性网络来识别四种主要组织之一特有的反式eQTL来验证这些发现。