Bernardes Juliana, Zaverucha Gerson, Vaquero Catherine, Carbone Alessandra
Sorbonne Universités, UPMC Univ-Paris 6, CNRS, UMR 7238, Laboratoire de Biologie Computationnelle et Quantitative, Paris, France.
COPPE, Programa de Engenharia de Sistemas e Computação, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil.
PLoS Comput Biol. 2016 Jul 29;12(7):e1005038. doi: 10.1371/journal.pcbi.1005038. eCollection 2016 Jul.
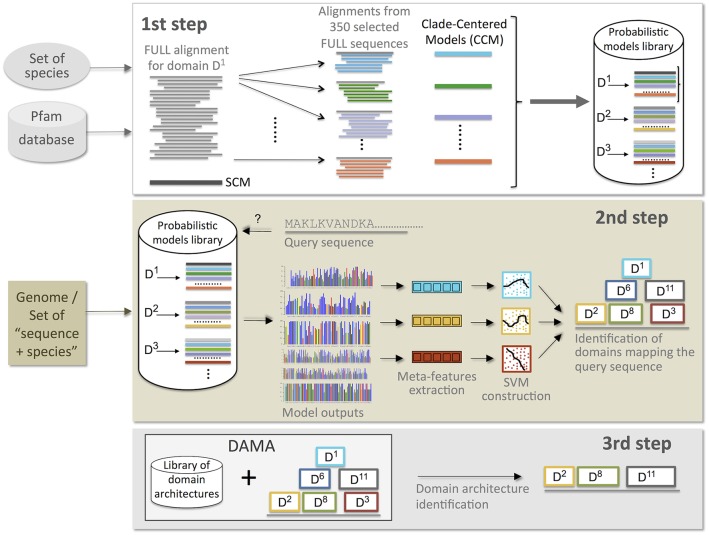
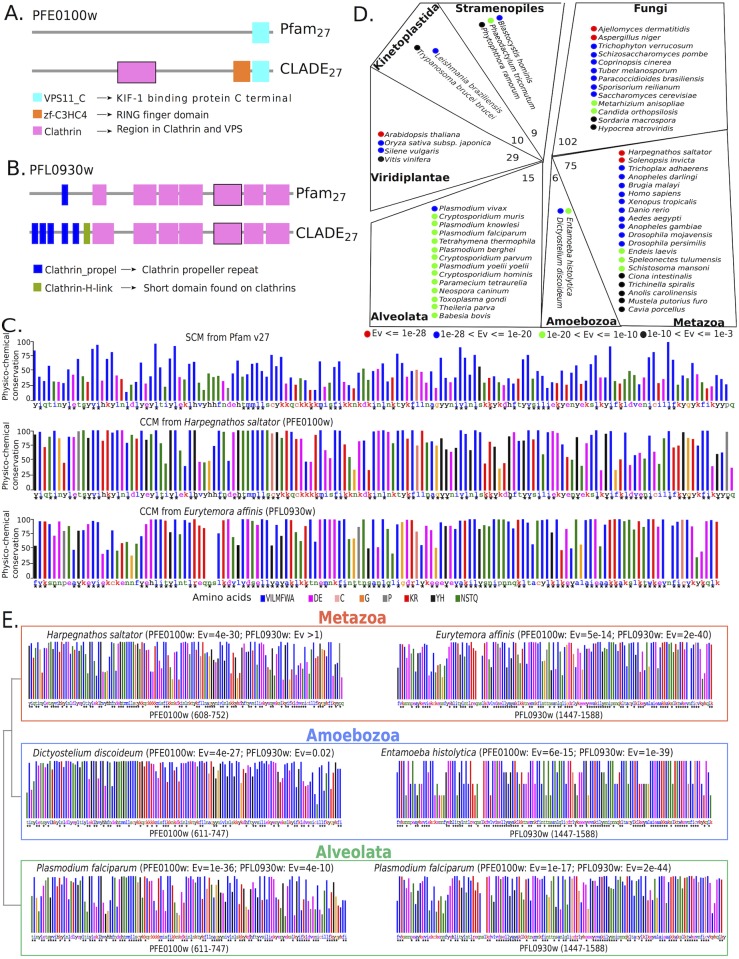
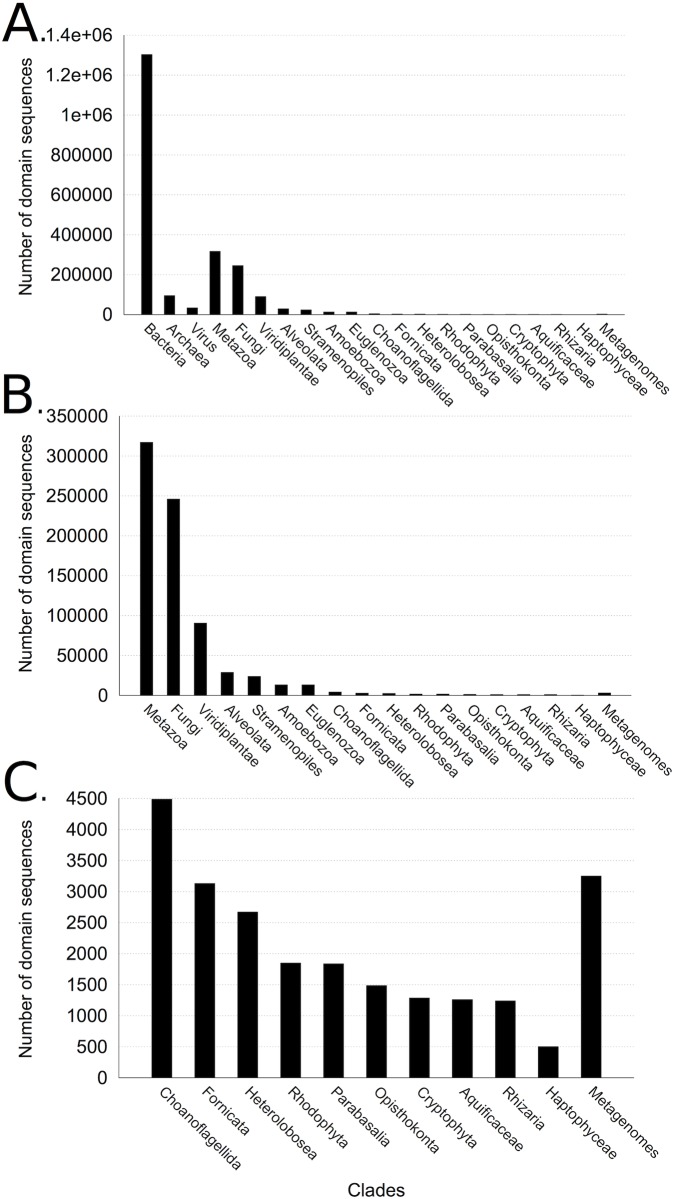
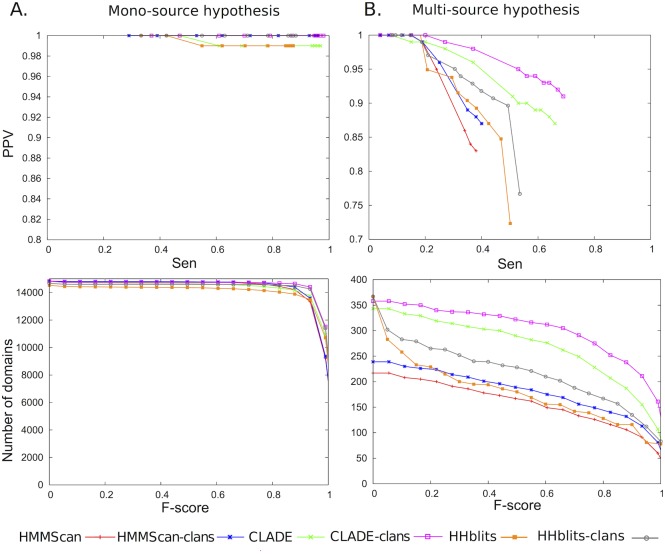
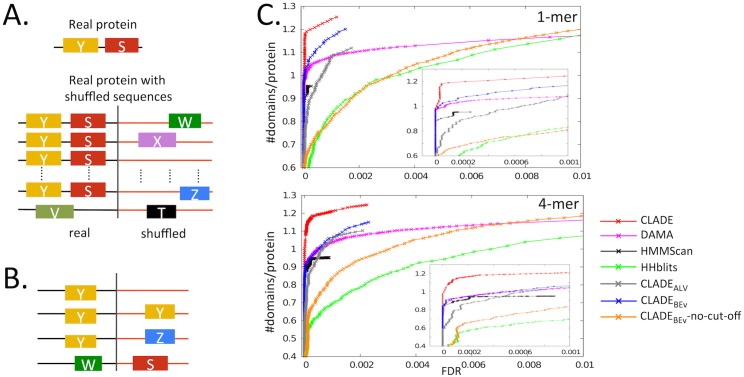
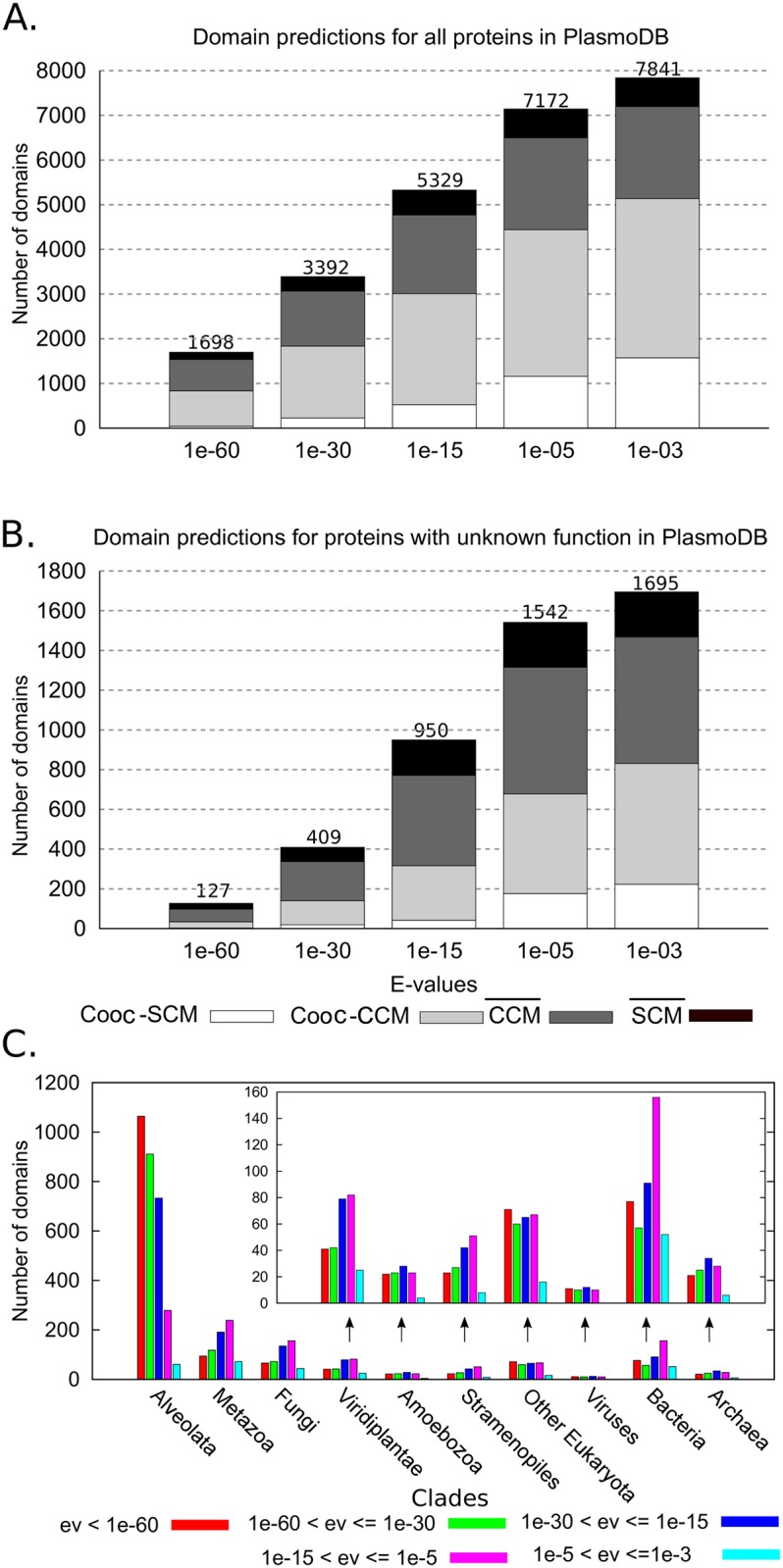
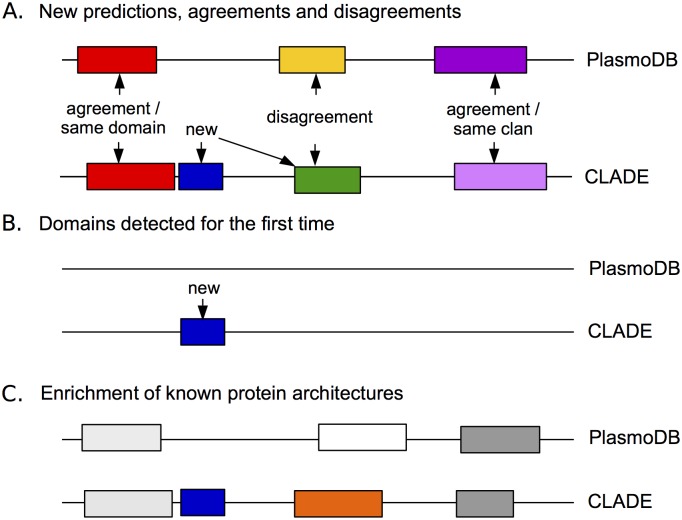
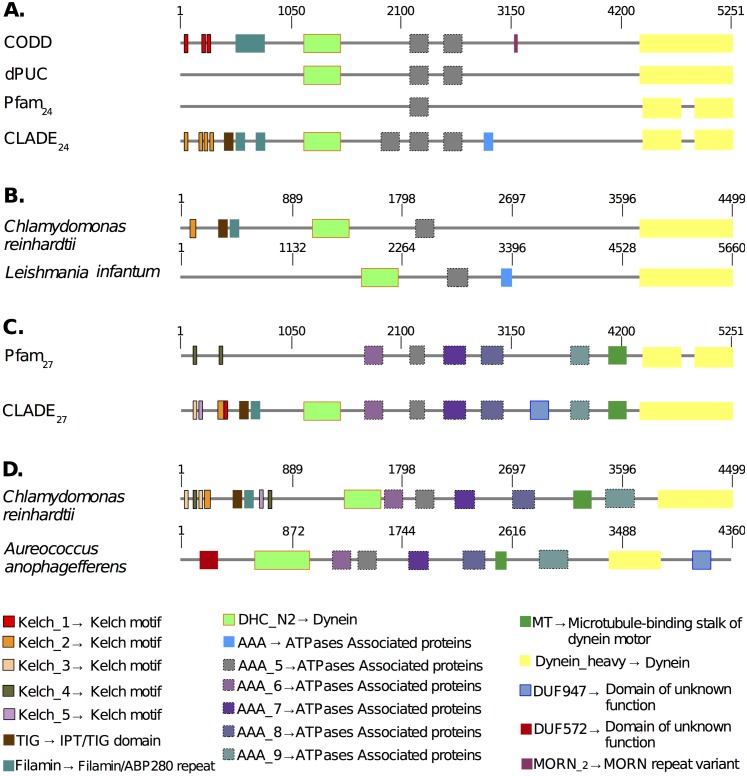
Traditional protein annotation methods describe known domains with probabilistic models representing consensus among homologous domain sequences. However, when relevant signals become too weak to be identified by a global consensus, attempts for annotation fail. Here we address the fundamental question of domain identification for highly divergent proteins. By using high performance computing, we demonstrate that the limits of state-of-the-art annotation methods can be bypassed. We design a new strategy based on the observation that many structural and functional protein constraints are not globally conserved through all species but might be locally conserved in separate clades. We propose a novel exploitation of the large amount of data available: 1. for each known protein domain, several probabilistic clade-centered models are constructed from a large and differentiated panel of homologous sequences, 2. a decision-making protocol combines outcomes obtained from multiple models, 3. a multi-criteria optimization algorithm finds the most likely protein architecture. The method is evaluated for domain and architecture prediction over several datasets and statistical testing hypotheses. Its performance is compared against HMMScan and HHblits, two widely used search methods based on sequence-profile and profile-profile comparison. Due to their closeness to actual protein sequences, clade-centered models are shown to be more specific and functionally predictive than the broadly used consensus models. Based on them, we improved annotation of Plasmodium falciparum protein sequences on a scale not previously possible. We successfully predict at least one domain for 72% of P. falciparum proteins against 63% achieved previously, corresponding to 30% of improvement over the total number of Pfam domain predictions on the whole genome. The method is applicable to any genome and opens new avenues to tackle evolutionary questions such as the reconstruction of ancient domain duplications, the reconstruction of the history of protein architectures, and the estimation of protein domain age. Website and software: http://www.lcqb.upmc.fr/CLADE.
传统的蛋白质注释方法使用概率模型来描述已知结构域,这些模型代表同源结构域序列之间的一致性。然而,当相关信号变得过于微弱以至于无法通过全局一致性来识别时,注释尝试就会失败。在这里,我们解决了高度分化蛋白质的结构域识别这一基本问题。通过使用高性能计算,我们证明了可以绕过现有最先进注释方法的局限性。我们基于这样的观察设计了一种新策略:许多蛋白质的结构和功能限制并非在所有物种中都全局保守,但可能在不同的进化枝中局部保守。我们提出了一种对可用大量数据的新颖利用方式:1. 对于每个已知的蛋白质结构域,从大量不同的同源序列组中构建几个以进化枝为中心的概率模型;2. 一个决策协议将从多个模型获得的结果结合起来;3. 一种多标准优化算法找到最可能的蛋白质结构。该方法在多个数据集上进行了结构域和结构预测以及统计检验假设的评估。其性能与HMMScan和HHblits这两种基于序列-轮廓和轮廓-轮廓比较的广泛使用的搜索方法进行了比较。由于以进化枝为中心的模型与实际蛋白质序列更接近,因此显示出比广泛使用的一致性模型更具特异性和功能预测性。基于这些模型,我们以前所未有的规模改进了恶性疟原虫蛋白质序列的注释。我们成功地为72%的恶性疟原虫蛋白质预测了至少一个结构域,而之前的成功率为63%,相对于全基因组上Pfam结构域预测总数提高了30%。该方法适用于任何基因组,并为解决进化问题开辟了新途径,如古代结构域复制的重建、蛋白质结构历史的重建以及蛋白质结构域年龄的估计。网站和软件:http://www.lcqb.upmc.fr/CLADE