Menichelli Christophe, Gascuel Olivier, Bréhélin Laurent
IBC, LIRMM, Univ. Montpellier, CNRS, Montpellier, France.
Unité de Bioinformatique Evolutive, C3BI - USR 3756, Institut Pasteur et CNRS, Paris, France.
PLoS Comput Biol. 2018 Jan 2;14(1):e1005889. doi: 10.1371/journal.pcbi.1005889. eCollection 2018 Jan.
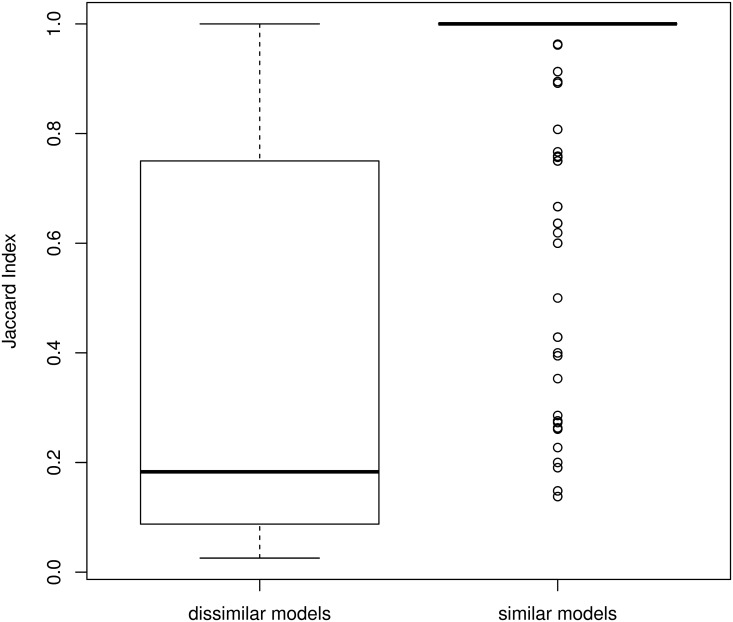
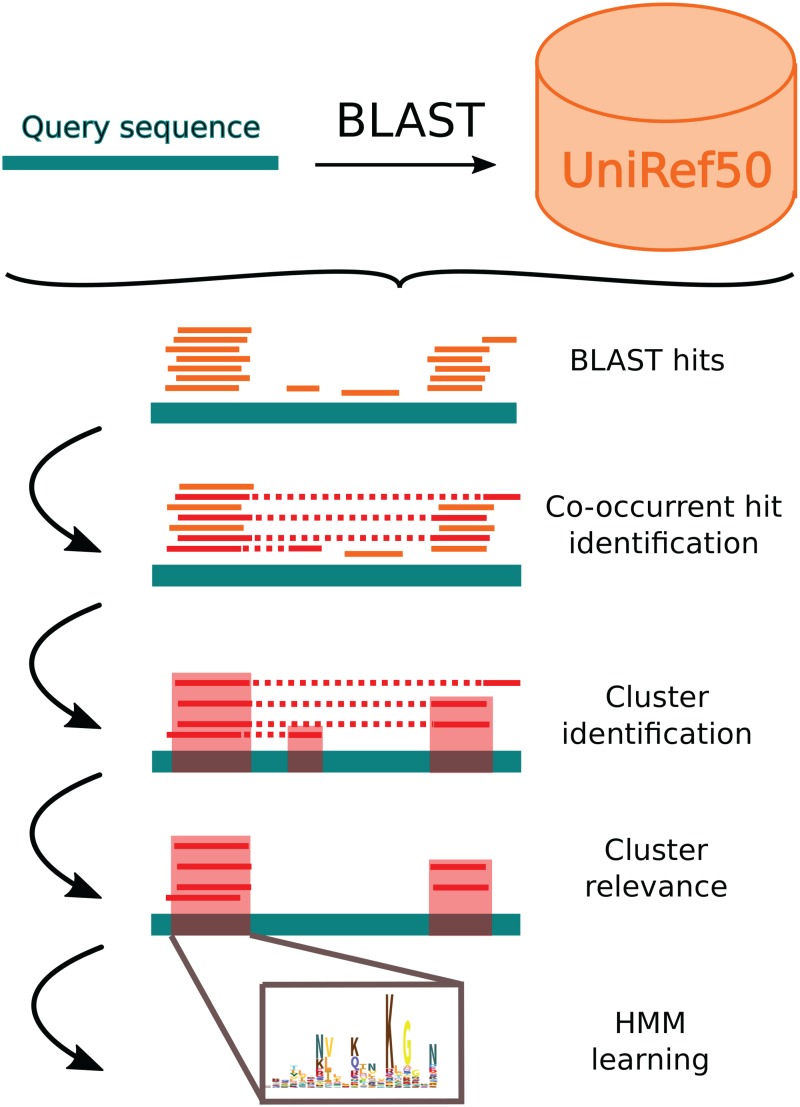
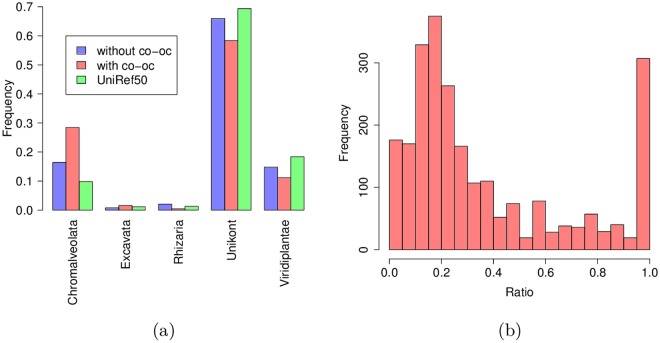
Comparing and aligning protein sequences is an essential task in bioinformatics. More specifically, local alignment tools like BLAST are widely used for identifying conserved protein sub-sequences, which likely correspond to protein domains or functional motifs. However, to limit the number of false positives, these tools are used with stringent sequence-similarity thresholds and hence can miss several hits, especially for species that are phylogenetically distant from reference organisms. A solution to this problem is then to integrate additional contextual information to the procedure. Here, we propose to use domain co-occurrence to increase the sensitivity of pairwise sequence comparisons. Domain co-occurrence is a strong feature of proteins, since most protein domains tend to appear with a limited number of other domains on the same protein. We propose a method to take this information into account in a typical BLAST analysis and to construct new domain families on the basis of these results. We used Plasmodium falciparum as a case study to evaluate our method. The experimental findings showed an increase of 14% of the number of significant BLAST hits and an increase of 25% of the proteome area that can be covered with a domain. Our method identified 2240 new domains for which, in most cases, no model of the Pfam database could be linked. Moreover, our study of the quality of the new domains in terms of alignment and physicochemical properties show that they are close to that of standard Pfam domains. Source code of the proposed approach and supplementary data are available at: https://gite.lirmm.fr/menichelli/pairwise-comparison-with-cooccurrence.
比较和比对蛋白质序列是生物信息学中的一项基本任务。更具体地说,像BLAST这样的局部比对工具被广泛用于识别保守的蛋白质子序列,这些子序列可能对应于蛋白质结构域或功能基序。然而,为了限制假阳性的数量,这些工具在使用时设置了严格的序列相似性阈值,因此可能会错过一些匹配结果,特别是对于那些在系统发育上与参考生物体距离较远的物种。解决这个问题的一个办法是将额外的上下文信息整合到这个过程中。在这里,我们建议使用结构域共现来提高成对序列比较的灵敏度。结构域共现是蛋白质的一个重要特征,因为大多数蛋白质结构域倾向于与同一蛋白质上数量有限的其他结构域一起出现。我们提出了一种方法,在典型的BLAST分析中考虑这些信息,并基于这些结果构建新的结构域家族。我们以恶性疟原虫为例来评估我们的方法。实验结果表明,显著的BLAST匹配结果数量增加了14%,蛋白质组中可以被一个结构域覆盖的区域增加了25%。我们的方法识别出了2240个新的结构域,在大多数情况下,这些结构域与Pfam数据库的模型没有关联。此外,我们对新结构域在比对和物理化学性质方面的质量研究表明,它们与标准的Pfam结构域相近。所提出方法的源代码和补充数据可在以下网址获取:https://gite.lirmm.fr/menichelli/pairwise-comparison-with-cooccurrence 。