Gianola Daniel, Schön Chris-Carolin
Department of Animal Sciences, University of Wisconsin-Madison, Wisconsin 53706 Department of Dairy Science, University of Wisconsin-Madison, Wisconsin 53706 Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Wisconsin 53706 Department of Plant Sciences, Technical University of Munich School of Life Sciences, Technical University of Munich, Garching, Germany Institute of Advanced Study, Technical University of Munich, Garching, Germany
Department of Plant Sciences, Technical University of Munich School of Life Sciences, Technical University of Munich, Garching, Germany Institute of Advanced Study, Technical University of Munich, Garching, Germany.
G3 (Bethesda). 2016 Oct 13;6(10):3107-3128. doi: 10.1534/g3.116.033381.
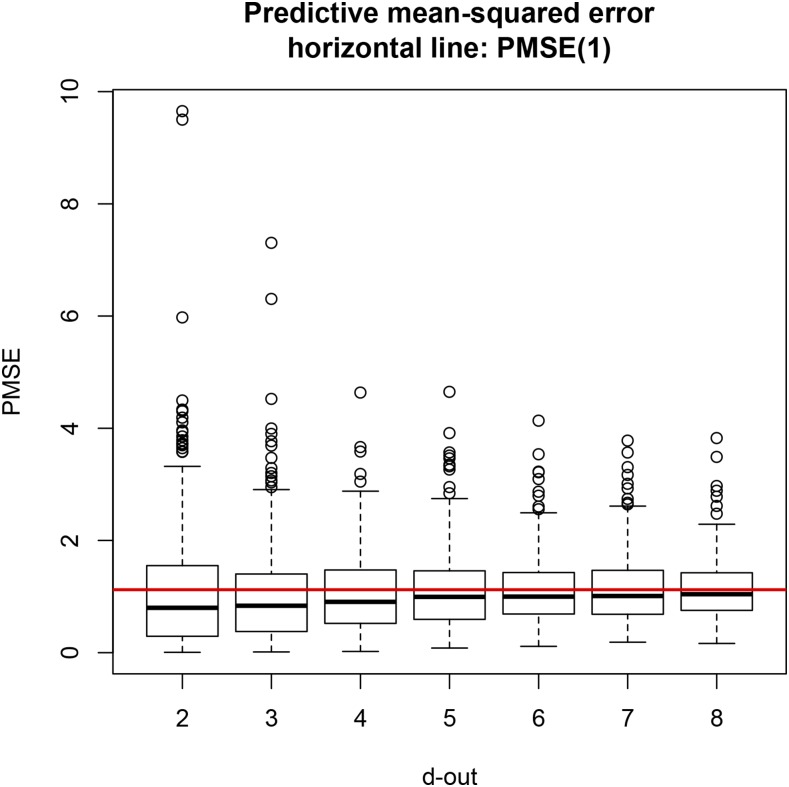
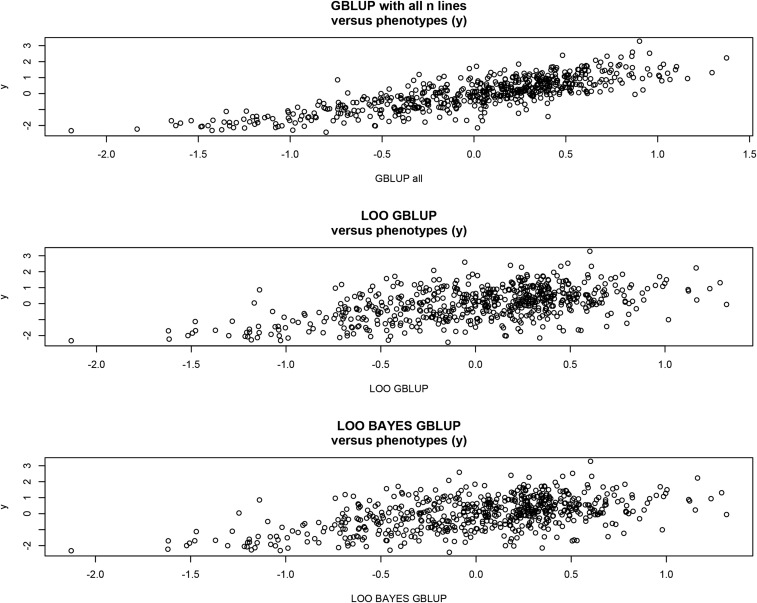
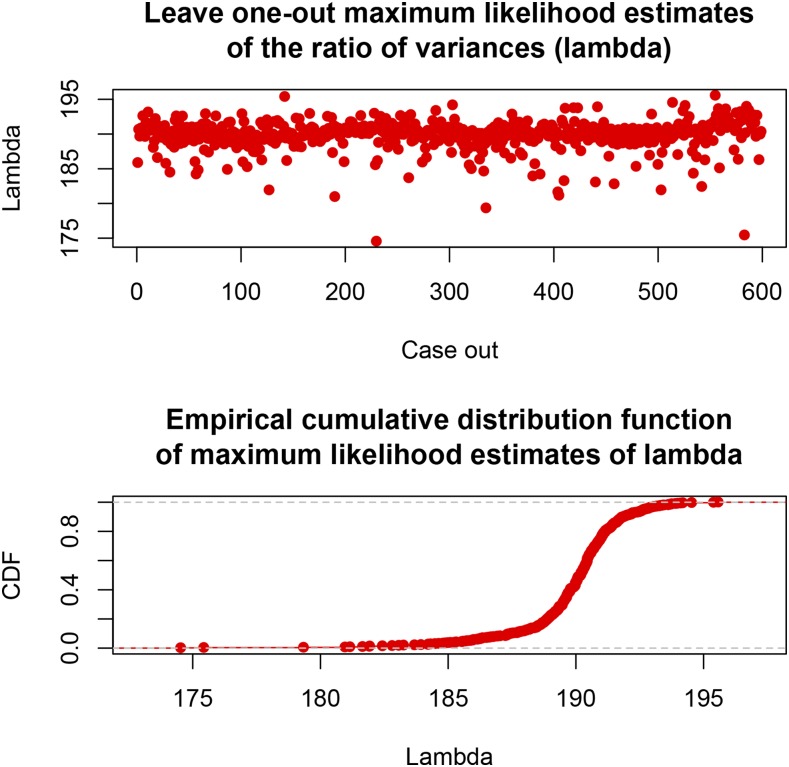
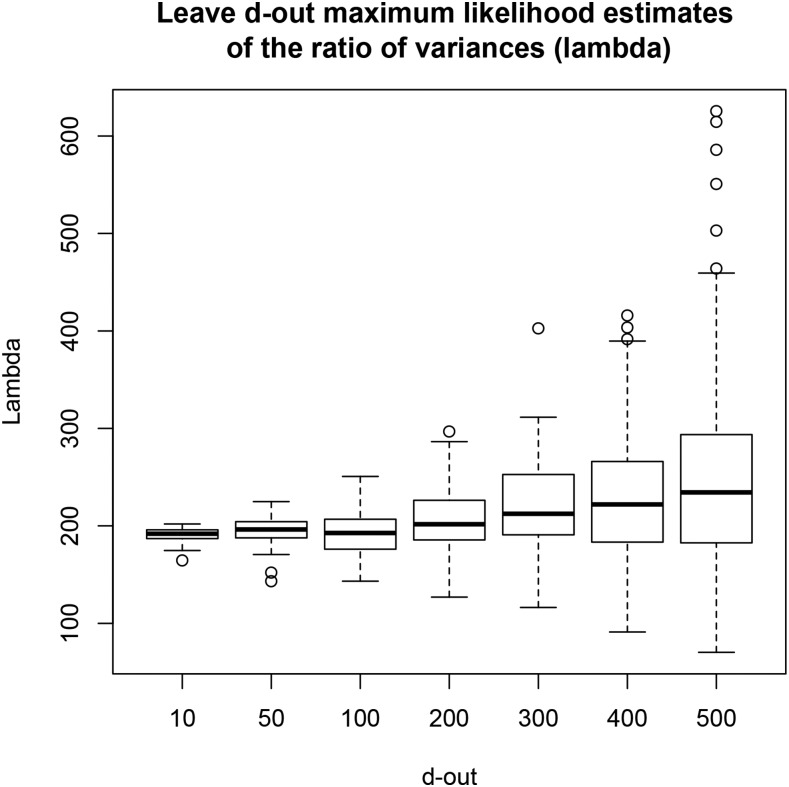
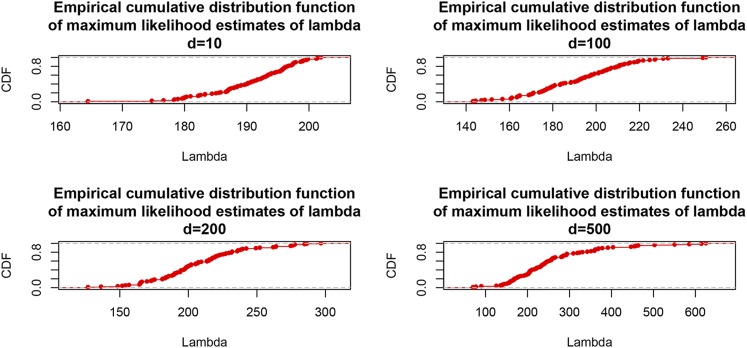
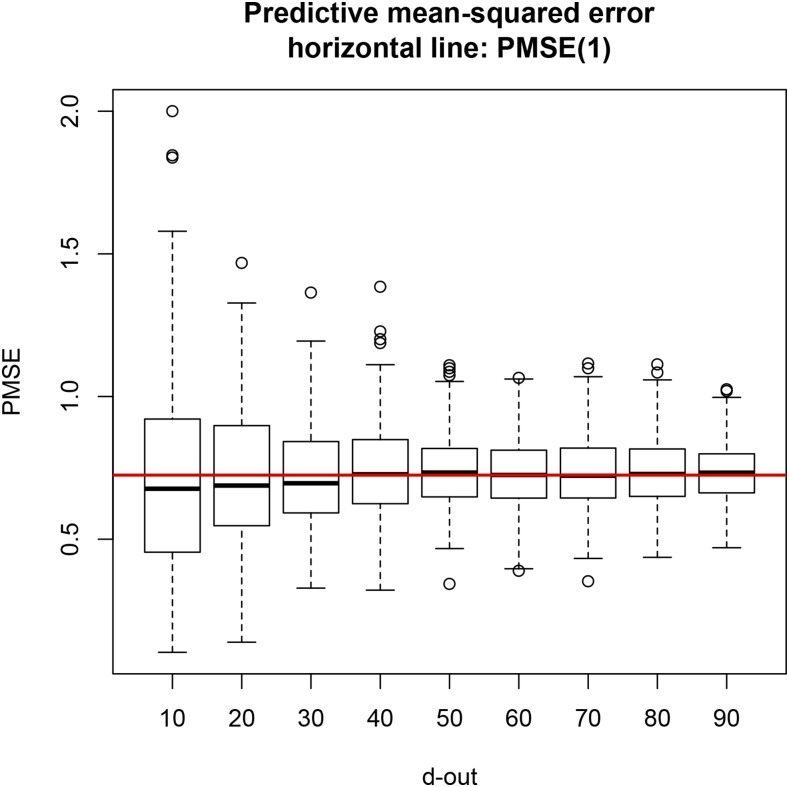
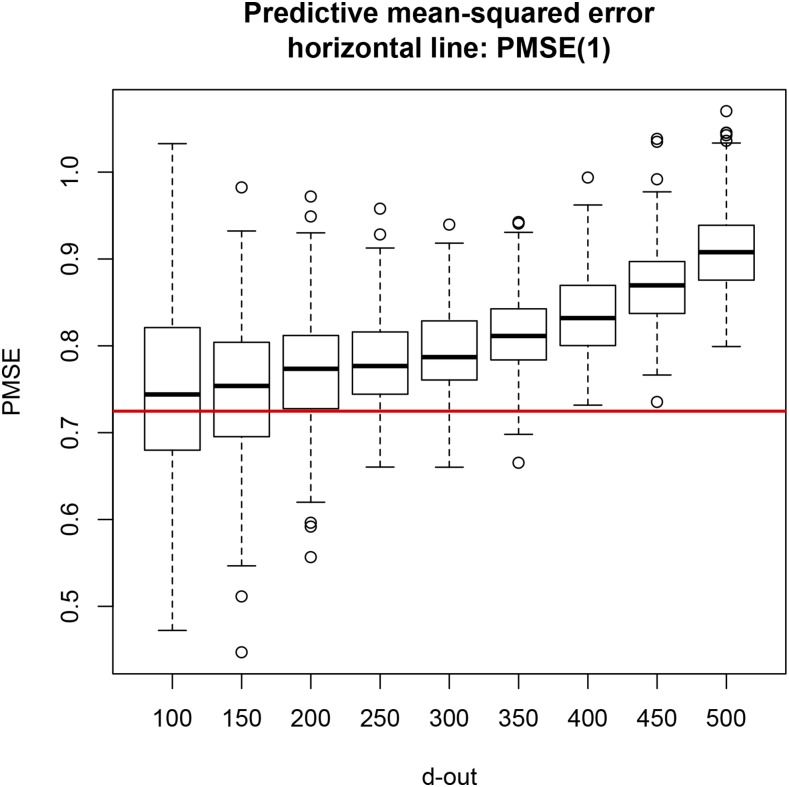
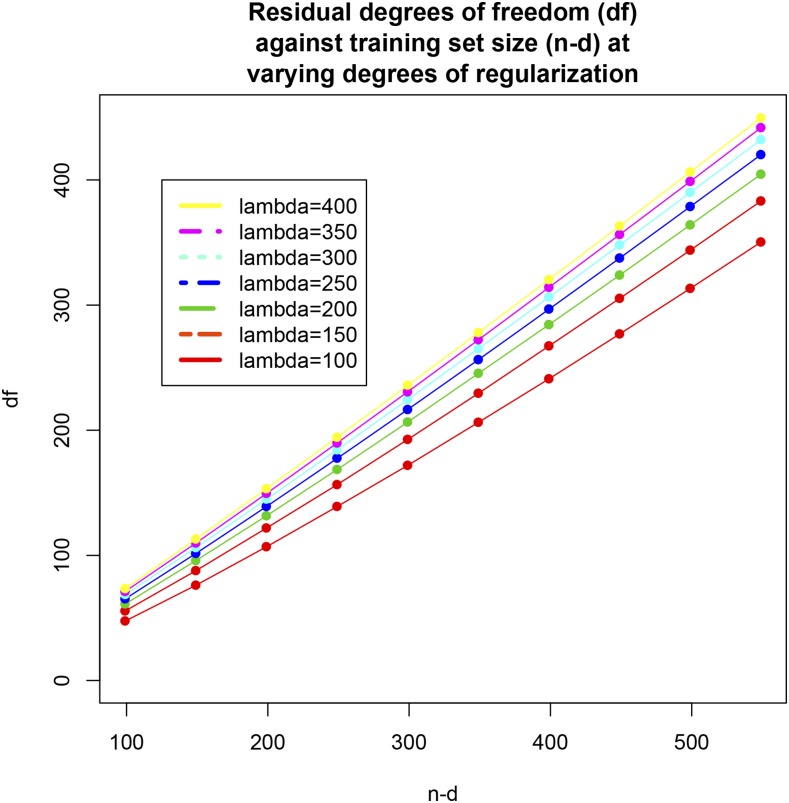
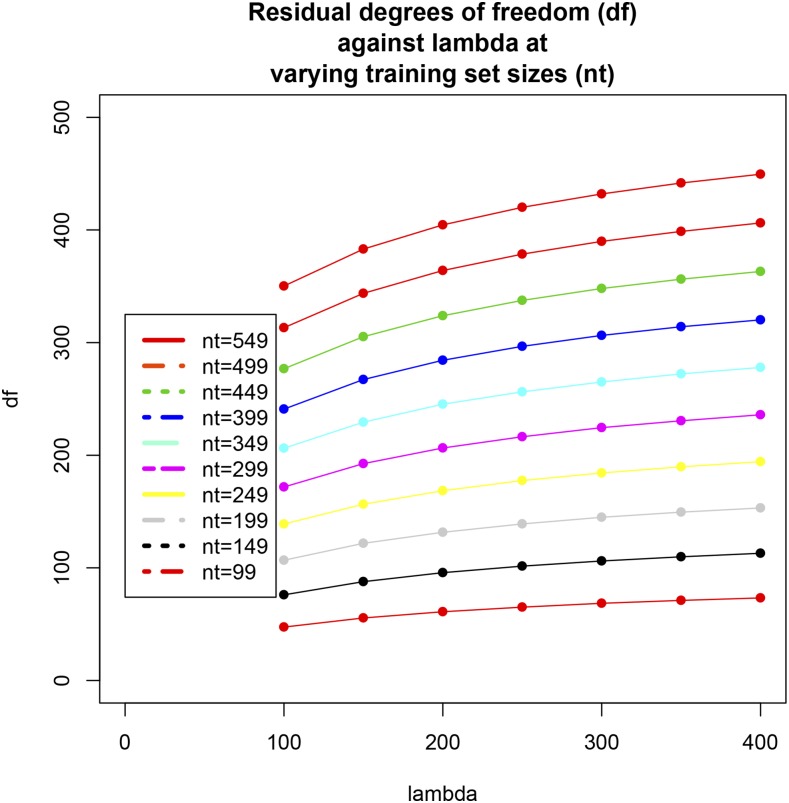
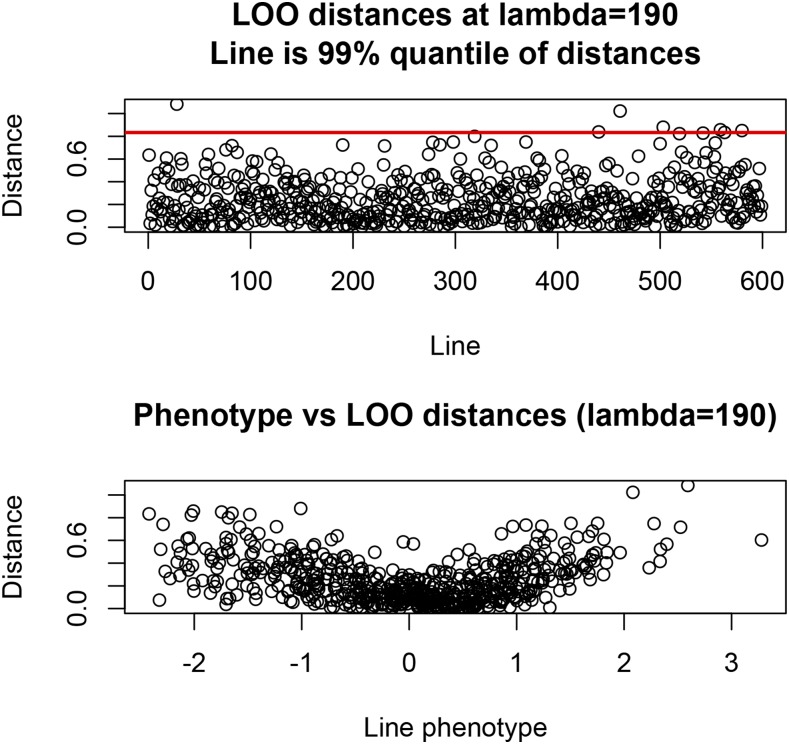
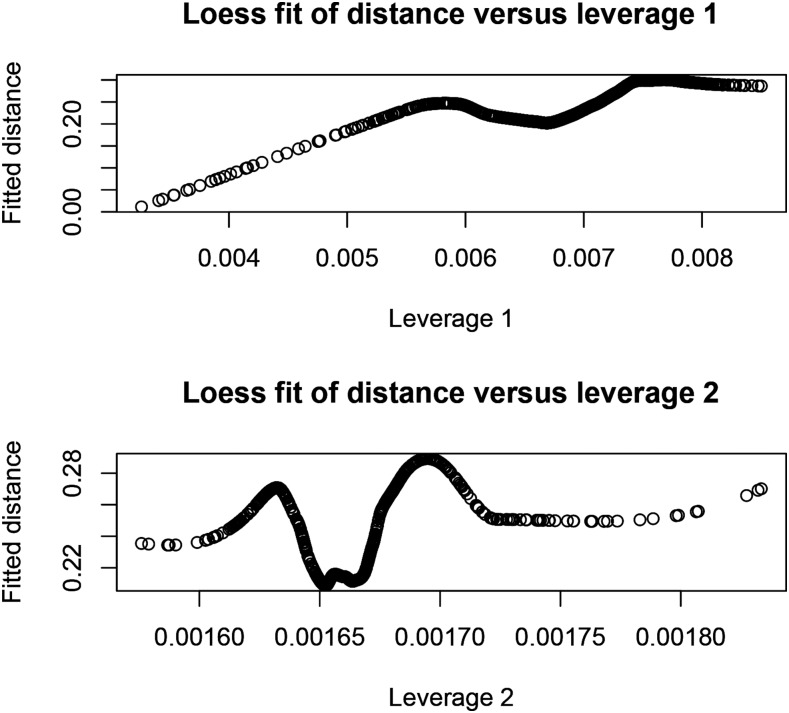
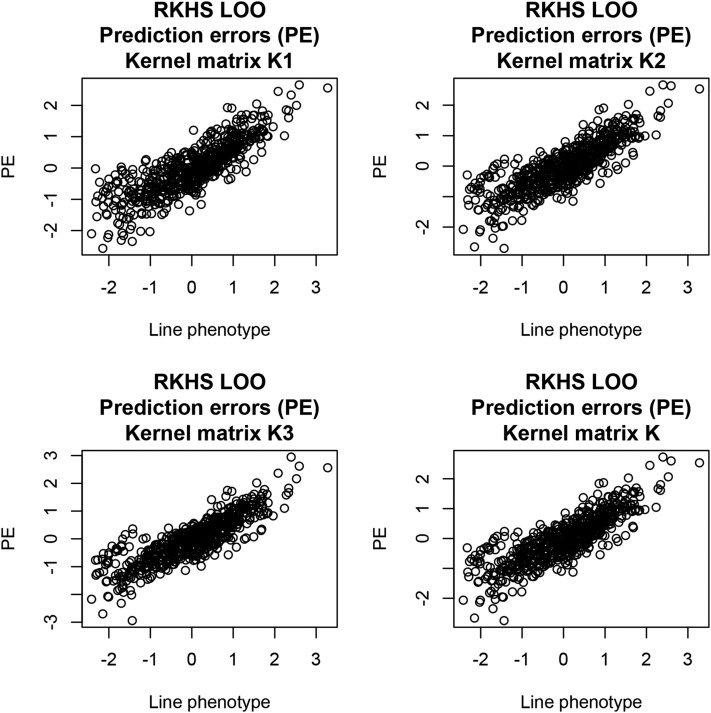
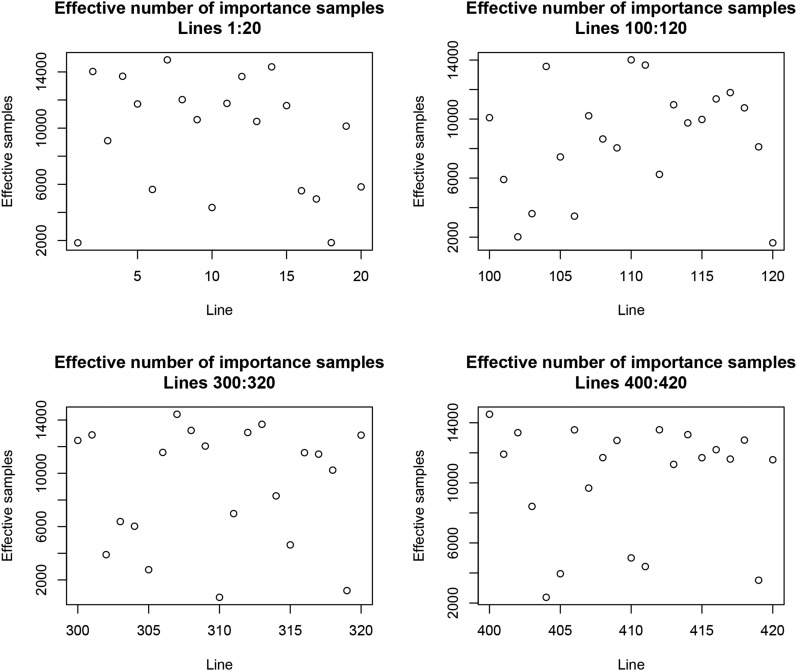
Cross-validation of methods is an essential component of genome-enabled prediction of complex traits. We develop formulae for computing the predictions that would be obtained when one or several cases are removed in the training process, to become members of testing sets, but by running the model using all observations only once. Prediction methods to which the developments apply include least squares, best linear unbiased prediction (BLUP) of markers, or genomic BLUP, reproducing kernels Hilbert spaces regression with single or multiple kernel matrices, and any member of a suite of linear regression methods known as "Bayesian alphabet." The approach used for Bayesian models is based on importance sampling of posterior draws. Proof of concept is provided by applying the formulae to a wheat data set representing 599 inbred lines genotyped for 1279 markers, and the target trait was grain yield. The data set was used to evaluate predictive mean-squared error, impact of alternative layouts on maximum likelihood estimates of regularization parameters, model complexity, and residual degrees of freedom stemming from various strengths of regularization, as well as two forms of importance sampling. Our results will facilitate carrying out extensive cross-validation without model retraining for most machines employed in genome-assisted prediction of quantitative traits.
方法的交叉验证是基于基因组的复杂性状预测的重要组成部分。我们开发了一些公式,用于计算在训练过程中移除一个或多个样本使其成为测试集成员,但仅使用所有观测值运行模型一次时所获得的预测结果。这些开发成果所适用的预测方法包括最小二乘法、标记的最佳线性无偏预测(BLUP)或基因组BLUP、使用单个或多个核矩阵的再生核希尔伯特空间回归,以及一组被称为“贝叶斯字母表”的线性回归方法中的任何一种。用于贝叶斯模型的方法基于后验抽样的重要性抽样。通过将这些公式应用于一个小麦数据集来提供概念验证,该数据集代表了599个自交系,对1279个标记进行了基因分型,目标性状是籽粒产量。该数据集用于评估预测均方误差、替代布局对正则化参数最大似然估计的影响、模型复杂性以及来自各种正则化强度的残差自由度,以及两种形式的重要性抽样。我们的结果将有助于在不进行模型重新训练的情况下,对大多数用于基因组辅助数量性状预测的机器进行广泛的交叉验证。