Favier Leslie A, Schulert Grant S
Division of Rheumatology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA.
Appl Clin Genet. 2016 Jul 20;9:101-10. doi: 10.2147/TACG.S93933. eCollection 2016.
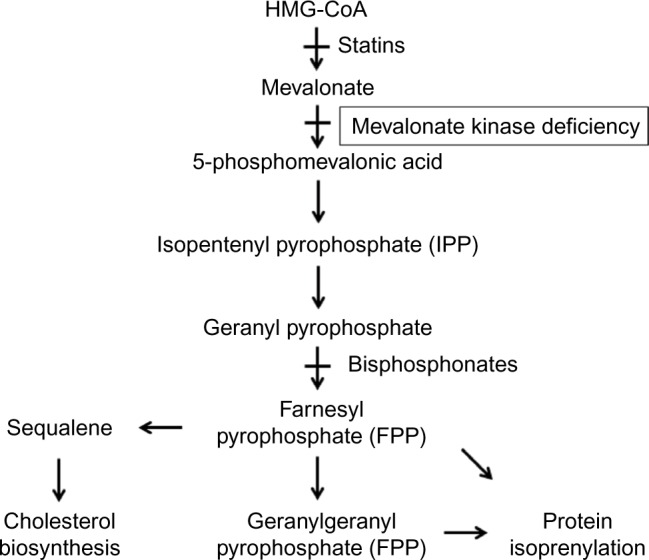
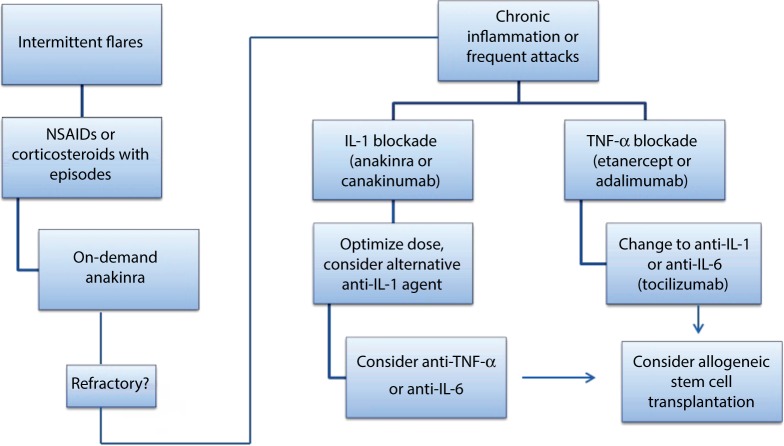
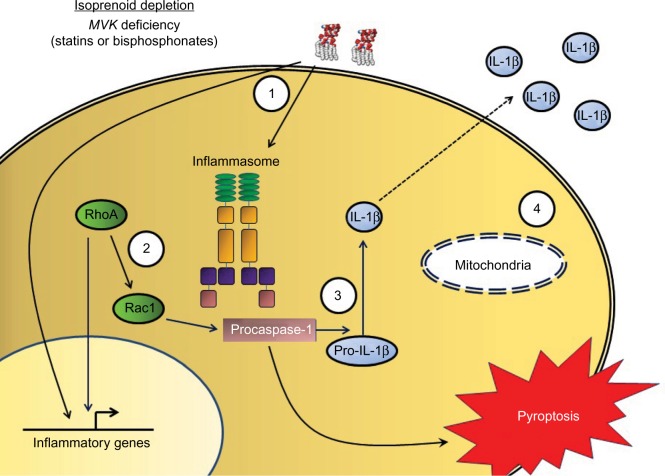
Mevalonate kinase deficiency (MKD) is a recessively inherited autoinflammatory disorder with a spectrum of manifestations, including the well-defined clinical phenotypes of hyperimmunoglobulinemia D and periodic fever syndrome and mevalonic aciduria. Patients with MKD have recurrent attacks of hyperinflammation associated with fever, abdominal pain, arthralgias, and mucocutaneous lesions, and more severely affected patients also have dysmorphisms and central nervous system anomalies. MKD is caused by mutations in the gene encoding mevalonate kinase, with the degree of residual enzyme activity largely determining disease severity. Mevalonate kinase is essential for the biosynthesis of nonsterol isoprenoids, which mediate protein prenylation. Although the precise pathogenesis of MKD remains unclear, increasing evidence suggests that deficiency in protein prenylation leads to innate immune activation and systemic hyperinflammation. Given the emerging understanding of MKD as an autoinflammatory disorder, recent treatment approaches have largely focused on cytokine-directed biologic therapy. Herein, we review the current genetic and pathologic understanding of MKD, its various clinical phenotypes, and the evolving treatment approach for this multifaceted disorder.
甲羟戊酸激酶缺乏症(MKD)是一种隐性遗传的自身炎症性疾病,具有一系列表现,包括明确的高免疫球蛋白D和周期性发热综合征以及甲羟戊酸尿症的临床表型。MKD患者会反复出现与发热、腹痛、关节痛和皮肤黏膜病变相关的高度炎症发作,病情更严重的患者还存在畸形和中枢神经系统异常。MKD是由编码甲羟戊酸激酶的基因突变引起的,残余酶活性的程度在很大程度上决定了疾病的严重程度。甲羟戊酸激酶对于非甾醇类异戊二烯的生物合成至关重要,而非甾醇类异戊二烯介导蛋白质异戊二烯化。尽管MKD的确切发病机制尚不清楚,但越来越多的证据表明,蛋白质异戊二烯化缺陷会导致先天性免疫激活和全身性高度炎症。鉴于对MKD作为一种自身炎症性疾病的认识不断深入,近期的治疗方法主要集中在细胞因子导向的生物治疗上。在此,我们综述了目前对MKD的遗传学和病理学认识、其各种临床表型以及针对这种多方面疾病不断演变的治疗方法。