Jungreis Irwin, Chan Clara S, Waterhouse Robert M, Fields Gabriel, Lin Michael F, Kellis Manolis
MIT Computer Science and Artificial Intelligence Laboratory, Cambridge, MA
Broad Institute of MIT and Harvard, Cambridge, MA.
Mol Biol Evol. 2016 Dec;33(12):3108-3132. doi: 10.1093/molbev/msw189. Epub 2016 Sep 7.
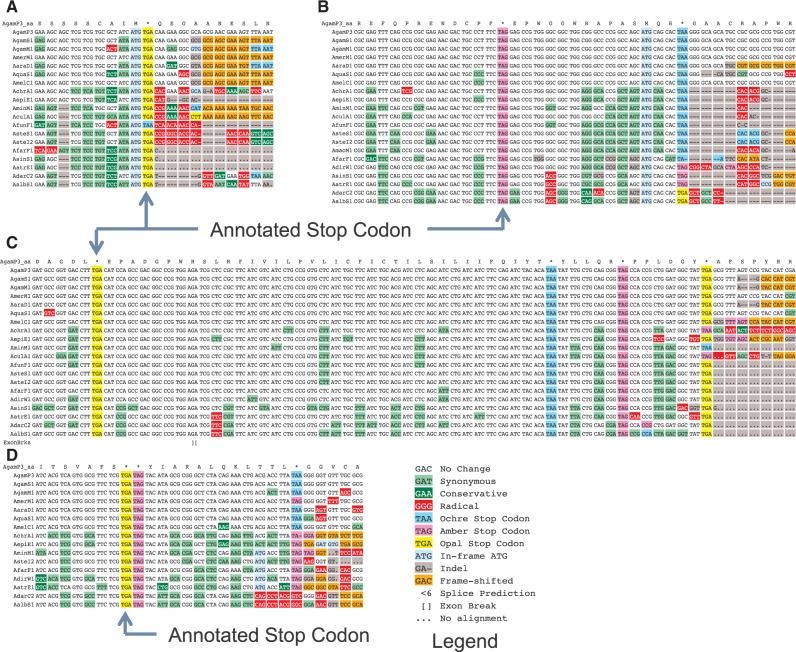
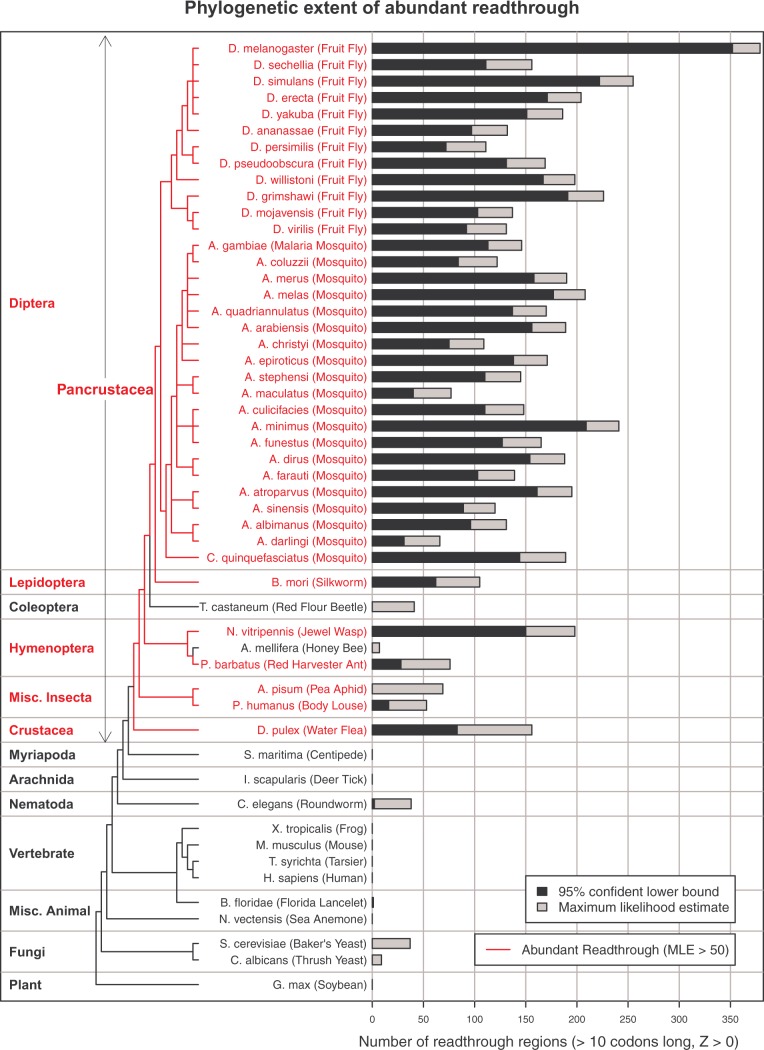
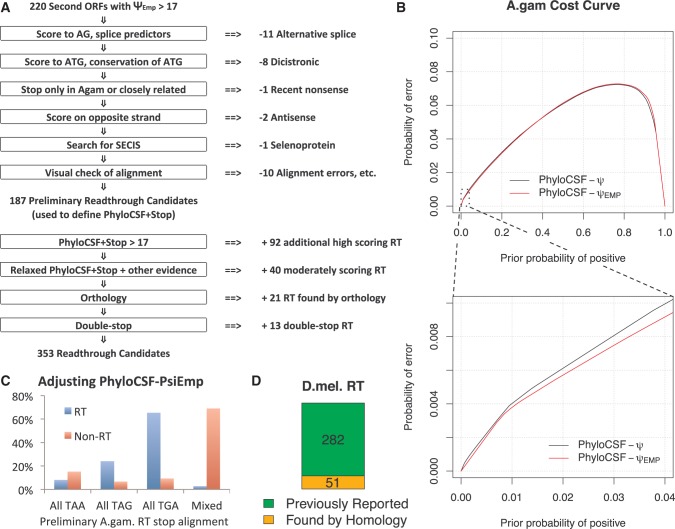
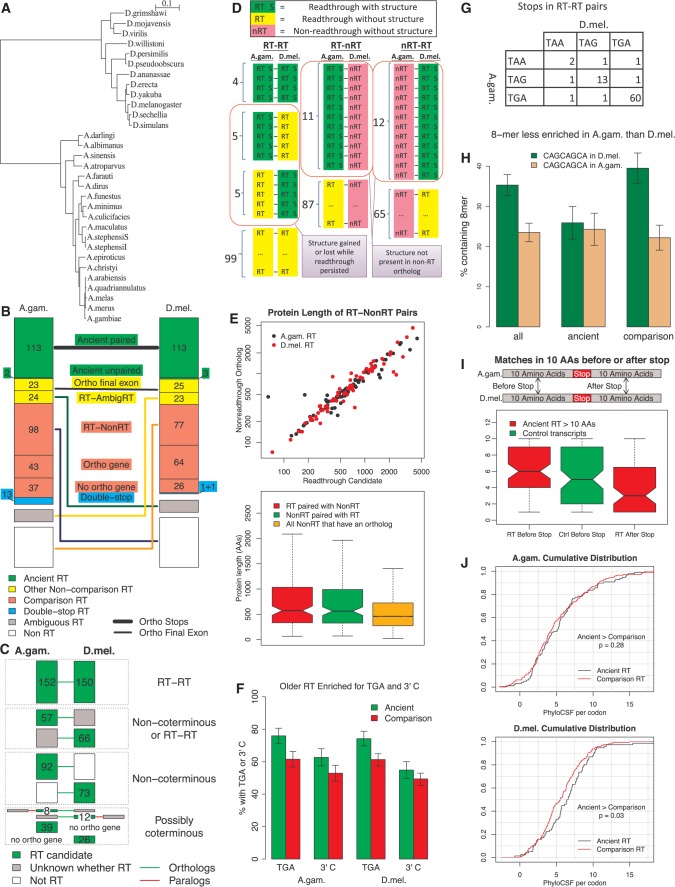
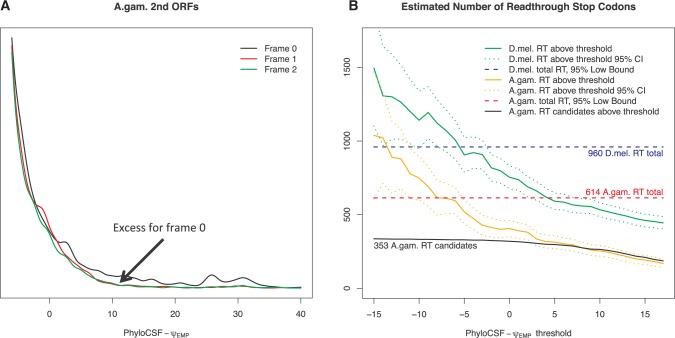
Translational stop codon readthrough emerged as a major regulatory mechanism affecting hundreds of genes in animal genomes, based on recent comparative genomics and ribosomal profiling evidence, but its evolutionary properties remain unknown. Here, we leverage comparative genomic evidence across 21 Anopheles mosquitoes to systematically annotate readthrough genes in the malaria vector Anopheles gambiae, and to provide the first study of abundant readthrough evolution, by comparison with 20 Drosophila species. Using improved comparative genomics methods for detecting readthrough, we identify evolutionary signatures of conserved, functional readthrough of 353 stop codons in the malaria vector, Anopheles gambiae, and of 51 additional Drosophila melanogaster stop codons, including several cases of double and triple readthrough and of readthrough of two adjacent stop codons. We find that most differences between the readthrough repertoires of the two species arose from readthrough gain or loss in existing genes, rather than birth of new genes or gene death; that readthrough-associated RNA structures are sometimes gained or lost while readthrough persists; that readthrough is more likely to be lost at TAA and TAG stop codons; and that readthrough is under continued purifying evolutionary selection in mosquito, based on population genetic evidence. We also determine readthrough-associated gene properties that predate readthrough, and identify differences in the characteristic properties of readthrough genes between clades. We estimate more than 600 functional readthrough stop codons in mosquito and 900 in fruit fly, provide evidence of readthrough control of peroxisomal targeting, and refine the phylogenetic extent of abundant readthrough as following divergence from centipede.
基于最近的比较基因组学和核糖体谱分析证据,翻译终止密码子通读已成为影响动物基因组中数百个基因的主要调控机制,但其进化特性仍不清楚。在这里,我们利用21种按蚊的比较基因组证据,系统地注释了疟疾媒介冈比亚按蚊中的通读基因,并通过与20种果蝇物种进行比较,首次对丰富的通读进化进行了研究。使用改进的比较基因组学方法来检测通读,我们在疟疾媒介冈比亚按蚊中鉴定出353个终止密码子保守、功能性通读的进化特征,以及另外51个黑腹果蝇终止密码子的进化特征,包括几例双重和三重通读以及两个相邻终止密码子的通读情况。我们发现,这两个物种通读库之间的大多数差异源于现有基因中通读的获得或丧失,而非新基因的产生或基因死亡;在通读持续存在的情况下,与通读相关的RNA结构有时会获得或丧失;通读在TAA和TAG终止密码子处更有可能丧失;并且基于群体遗传学证据,通读在蚊子中受到持续的纯化进化选择。我们还确定了早于通读的与通读相关的基因特性,并识别出不同进化枝之间通读基因特征特性的差异。我们估计蚊子中有600多个功能性通读终止密码子,果蝇中有900多个,提供了过氧化物酶体靶向通读控制的证据,并将丰富通读的系统发育范围细化为自与蜈蚣分化之后的情况。