Ray Bisakha, Ghedin Elodie, Chunara Rumi
Center for Health Informatics and Bioinformatics, New York University School of Medicine, USA.
Department of Biology, Center for Genomics & Systems Biology, USA; College of Global Public Health, New York University, USA.
J Biomed Inform. 2016 Dec;64:44-54. doi: 10.1016/j.jbi.2016.09.004. Epub 2016 Sep 6.
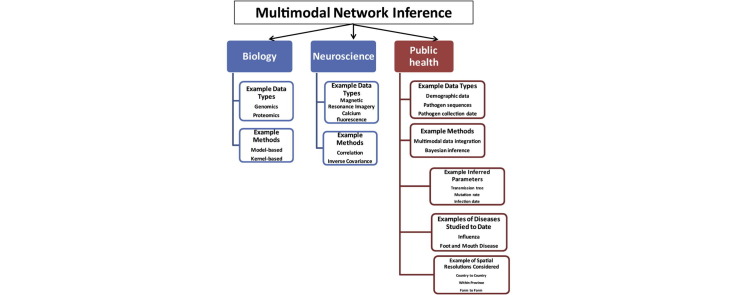
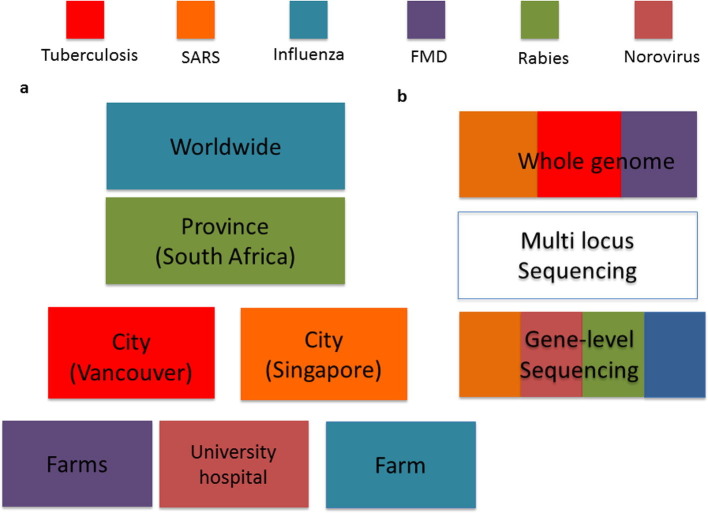
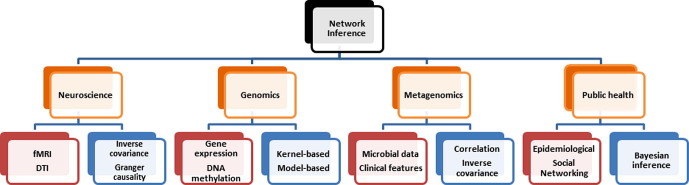
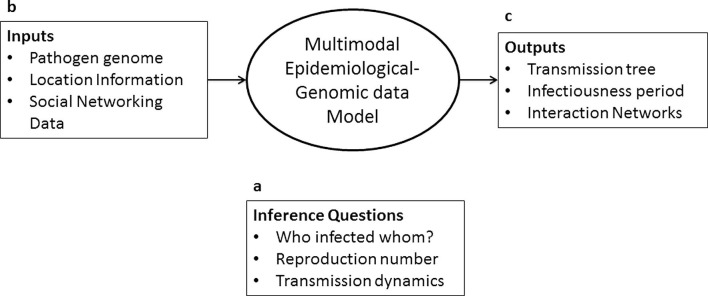
Networks inference problems are commonly found in multiple biomedical subfields such as genomics, metagenomics, neuroscience, and epidemiology. Networks are useful for representing a wide range of complex interactions ranging from those between molecular biomarkers, neurons, and microbial communities, to those found in human or animal populations. Recent technological advances have resulted in an increasing amount of healthcare data in multiple modalities, increasing the preponderance of network inference problems. Multi-domain data can now be used to improve the robustness and reliability of recovered networks from unimodal data. For infectious diseases in particular, there is a body of knowledge that has been focused on combining multiple pieces of linked information. Combining or analyzing disparate modalities in concert has demonstrated greater insight into disease transmission than could be obtained from any single modality in isolation. This has been particularly helpful in understanding incidence and transmission at early stages of infections that have pandemic potential. Novel pieces of linked information in the form of spatial, temporal, and other covariates including high-throughput sequence data, clinical visits, social network information, pharmaceutical prescriptions, and clinical symptoms (reported as free-text data) also encourage further investigation of these methods. The purpose of this review is to provide an in-depth analysis of multimodal infectious disease transmission network inference methods with a specific focus on Bayesian inference. We focus on analytical Bayesian inference-based methods as this enables recovering multiple parameters simultaneously, for example, not just the disease transmission network, but also parameters of epidemic dynamics. Our review studies their assumptions, key inference parameters and limitations, and ultimately provides insights about improving future network inference methods in multiple applications.
网络推理问题常见于多个生物医学子领域,如基因组学、宏基因组学、神经科学和流行病学。网络有助于表示广泛的复杂相互作用,从分子生物标志物、神经元和微生物群落之间的相互作用,到人类或动物群体中的相互作用。最近的技术进步导致了多种形式的医疗保健数据量不断增加,从而增加了网络推理问题的比重。现在,多领域数据可用于提高从单峰数据中恢复的网络的稳健性和可靠性。特别是对于传染病,已有大量知识专注于整合多条相关信息。协同组合或分析不同形式的数据已证明,比起单独从任何一种形式的数据中所能获得的信息,这样能更深入地洞察疾病传播。这在理解具有大流行潜力的感染早期阶段的发病率和传播方面特别有帮助。以空间、时间和其他协变量形式呈现的新的相关信息,包括高通量序列数据、临床就诊记录、社交网络信息、药物处方和临床症状(以自由文本数据形式报告),也促使人们进一步研究这些方法。本综述的目的是对多模态传染病传播网络推理方法进行深入分析,特别关注贝叶斯推理。我们专注于基于分析贝叶斯推理的方法,因为这能够同时恢复多个参数,例如,不仅能恢复疾病传播网络,还能恢复疫情动态的参数。我们的综述研究了它们的假设、关键推理参数和局限性,并最终为在多种应用中改进未来的网络推理方法提供见解。