Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
Department of Ecology and Evolutionary Biology, Princeton University, Princeton, New Jersey, United States of America.
PLoS Comput Biol. 2018 Apr 18;14(4):e1006117. doi: 10.1371/journal.pcbi.1006117. eCollection 2018 Apr.
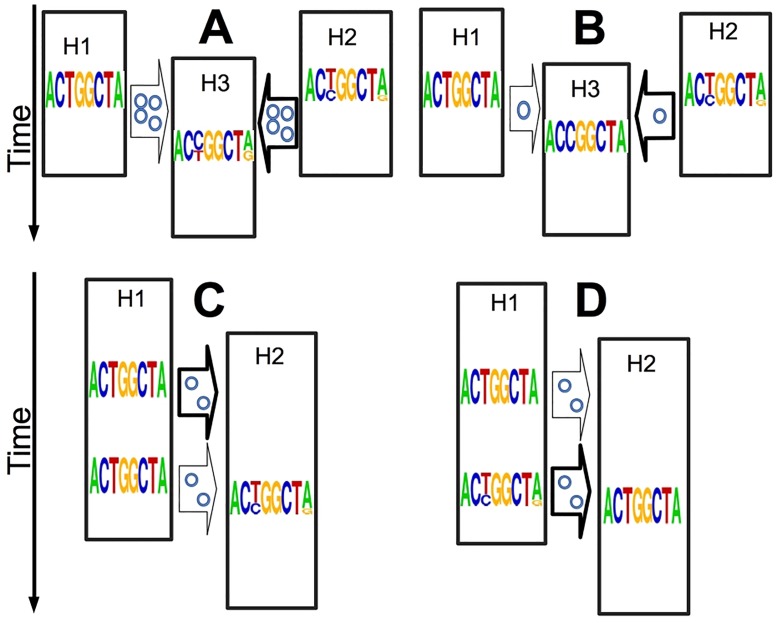
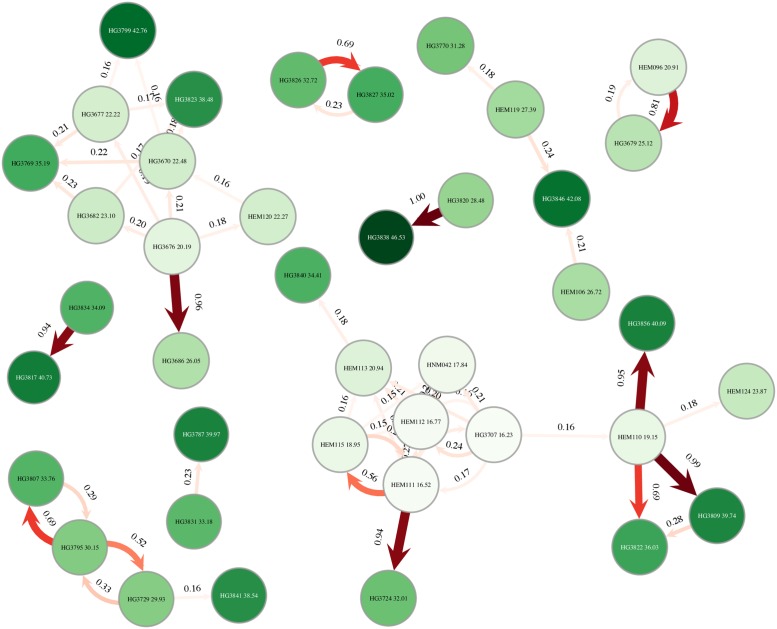
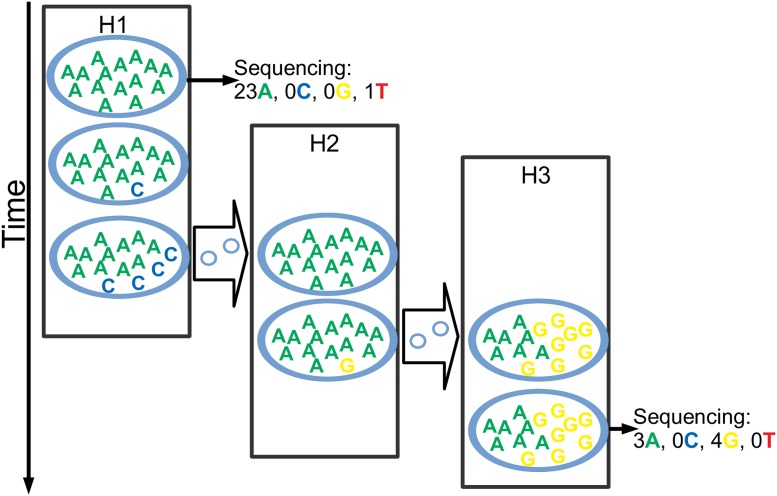
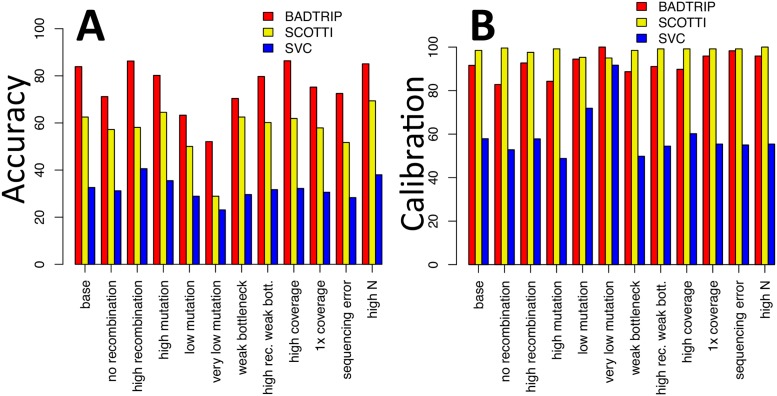
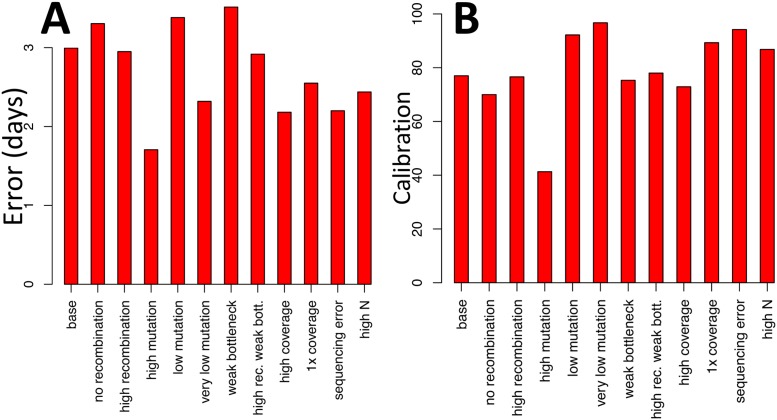
Pathogen genome sequencing can reveal details of transmission histories and is a powerful tool in the fight against infectious disease. In particular, within-host pathogen genomic variants identified through heterozygous nucleotide base calls are a potential source of information to identify linked cases and infer direction and time of transmission. However, using such data effectively to model disease transmission presents a number of challenges, including differentiating genuine variants from those observed due to sequencing error, as well as the specification of a realistic model for within-host pathogen population dynamics. Here we propose a new Bayesian approach to transmission inference, BadTrIP (BAyesian epiDemiological TRansmission Inference from Polymorphisms), that explicitly models evolution of pathogen populations in an outbreak, transmission (including transmission bottlenecks), and sequencing error. BadTrIP enables the inference of host-to-host transmission from pathogen sequencing data and epidemiological data. By assuming that genomic variants are unlinked, our method does not require the computationally intensive and unreliable reconstruction of individual haplotypes. Using simulations we show that BadTrIP is robust in most scenarios and can accurately infer transmission events by efficiently combining information from genetic and epidemiological sources; thanks to its realistic model of pathogen evolution and the inclusion of epidemiological data, BadTrIP is also more accurate than existing approaches. BadTrIP is distributed as an open source package (https://bitbucket.org/nicofmay/badtrip) for the phylogenetic software BEAST2. We apply our method to reconstruct transmission history at the early stages of the 2014 Ebola outbreak, showcasing the power of within-host genomic variants to reconstruct transmission events.
病原体基因组测序可以揭示传播史的细节,是对抗传染病的有力工具。特别是,通过杂合核苷酸碱基调用鉴定的宿主内病原体基因组变异体是识别关联病例并推断传播方向和时间的潜在信息来源。然而,有效地利用此类数据来模拟疾病传播存在许多挑战,包括区分真正的变异体和由于测序错误而观察到的变异体,以及为宿主内病原体种群动态指定现实的模型。在这里,我们提出了一种新的贝叶斯方法来进行传播推断,即 BadTrIP(基于多态性的贝叶斯流行病学传播推断),它明确地模拟了暴发中的病原体种群进化、传播(包括传播瓶颈)和测序错误。BadTrIP 能够从病原体测序数据和流行病学数据中推断宿主间的传播。通过假设基因组变异体是不相关的,我们的方法不需要对个体单倍型进行计算密集型且不可靠的重建。通过模拟,我们表明 BadTrIP 在大多数情况下是稳健的,可以通过有效地结合遗传和流行病学来源的信息来准确推断传播事件;由于其对病原体进化的现实模型和流行病学数据的纳入,BadTrIP 比现有方法更准确。BadTrIP 作为一个开源软件包(https://bitbucket.org/nicofmay/badtrip)分发给系统发育软件 BEAST2。我们应用我们的方法来重建 2014 年埃博拉疫情早期的传播历史,展示了宿主内基因组变异体重建传播事件的强大功能。