Yung Godwin, Lin Xihong
Department of Biostatistics, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, United States of America.
Genet Epidemiol. 2016 Dec;40(8):732-743. doi: 10.1002/gepi.21994. Epub 2016 Sep 26.
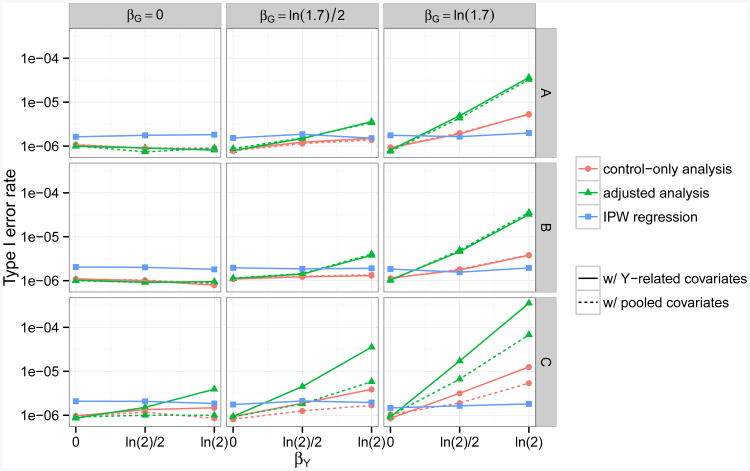
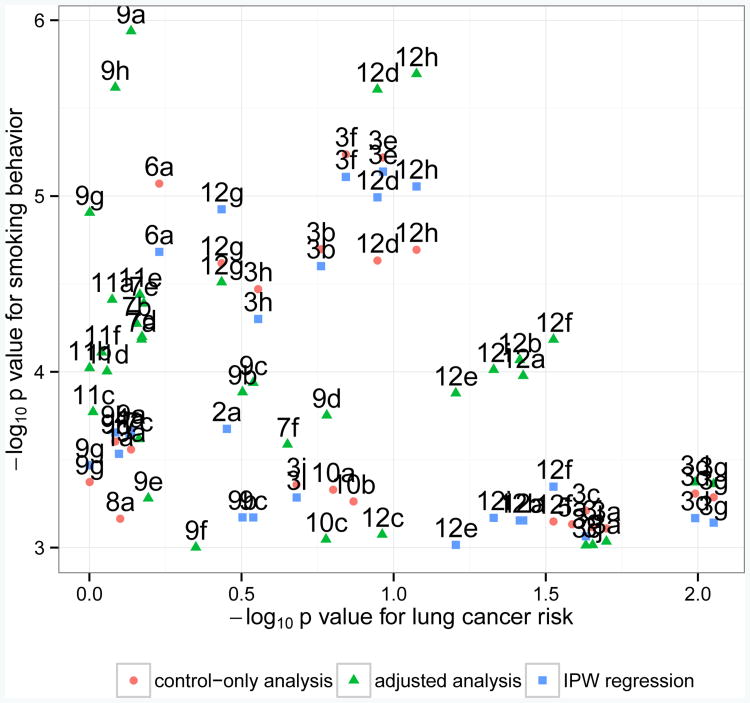
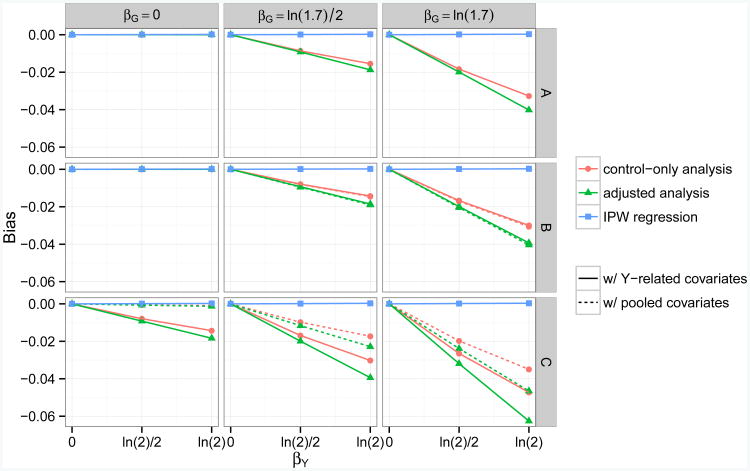
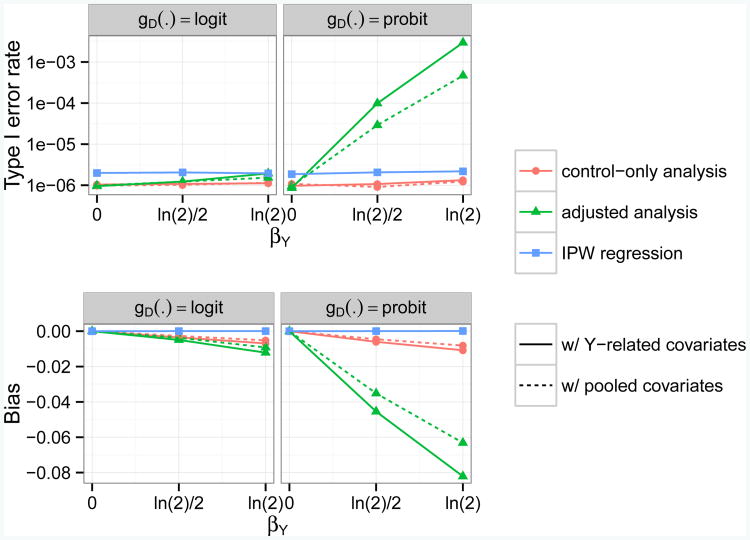
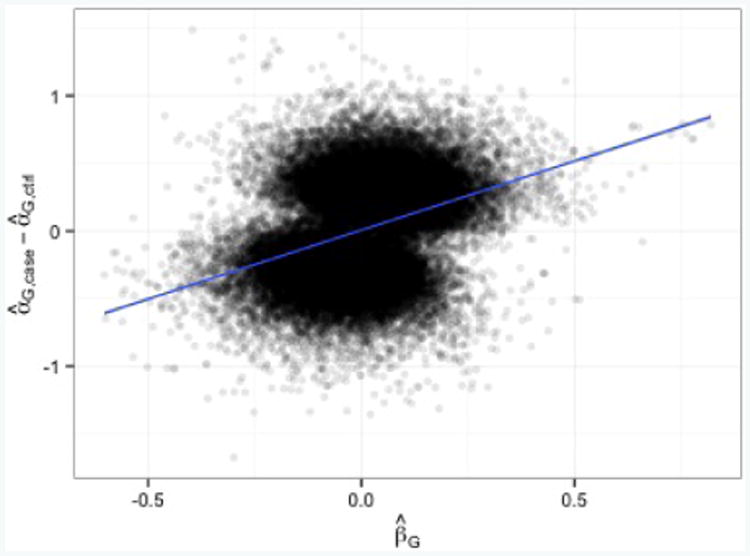
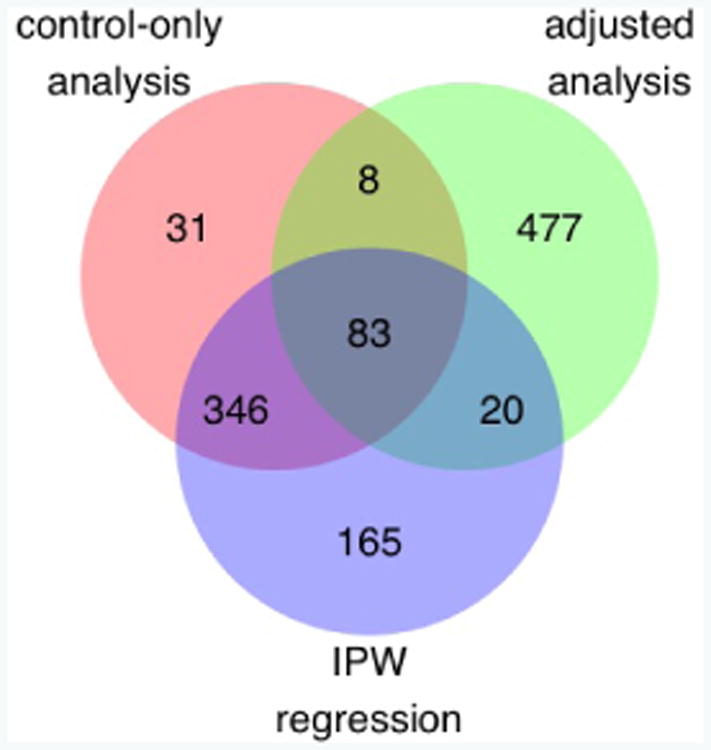
Case-control association studies often collect from their subjects information on secondary phenotypes. Reusing the data and studying the association between genes and secondary phenotypes provide an attractive and cost-effective approach that can lead to discovery of new genetic associations. A number of approaches have been proposed, including simple and computationally efficient ad hoc methods that ignore ascertainment or stratify on case-control status. Justification for these approaches relies on the assumption of no covariates and the correct specification of the primary disease model as a logistic model. Both might not be true in practice, for example, in the presence of population stratification or the primary disease model following a probit model. In this paper, we investigate the validity of ad hoc methods in the presence of covariates and possible disease model misspecification. We show that in taking an ad hoc approach, it may be desirable to include covariates that affect the primary disease in the secondary phenotype model, even though these covariates are not necessarily associated with the secondary phenotype. We also show that when the disease is rare, ad hoc methods can lead to severely biased estimation and inference if the true disease model follows a probit model instead of a logistic model. Our results are justified theoretically and via simulations. Applied to real data analysis of genetic associations with cigarette smoking, ad hoc methods collectively identified as highly significant (P<10-5) single nucleotide polymorphisms from over 10 genes, genes that were identified in previous studies of smoking cessation.
病例对照关联研究通常会收集其研究对象的次要表型信息。重新利用这些数据并研究基因与次要表型之间的关联,提供了一种有吸引力且具有成本效益的方法,可能会带来新的基因关联的发现。已经提出了许多方法,包括简单且计算效率高的临时方法,这些方法忽略了样本确定过程或按病例对照状态进行分层。这些方法的合理性依赖于无协变量的假设以及将原发性疾病模型正确设定为逻辑模型。在实际中这两者可能都不成立,例如,存在群体分层或原发性疾病模型遵循概率单位模型的情况。在本文中,我们研究了在存在协变量和可能的疾病模型错误设定的情况下临时方法的有效性。我们表明,采用临时方法时,在次要表型模型中纳入影响原发性疾病的协变量可能是可取的,即使这些协变量不一定与次要表型相关。我们还表明,当疾病罕见时,如果真实的疾病模型遵循概率单位模型而非逻辑模型,临时方法可能会导致严重有偏的估计和推断。我们的结果在理论上和通过模拟得到了验证。应用于与吸烟的基因关联的实际数据分析时,临时方法共同从超过10个基因中识别出了被确定为高度显著(P<10-5)的单核苷酸多态性,这些基因是在先前戒烟研究中被识别出的。