Abe Yoshifusa, Iyoda Masayuki, Nozu Kandai, Hibino Satoshi, Hihara Kei, Yamaguchi Yutaka, Yamamura Tomohiko, Minamikawa Shogo, Iijima Kazumoto, Shibata Takanori, Itabashi Kazuo
Department of Pediatrics, Showa University School of Medicine, Japan.
Intern Med. 2016;55(19):2843-2847. doi: 10.2169/internalmedicine.55.6873. Epub 2016 Oct 1.
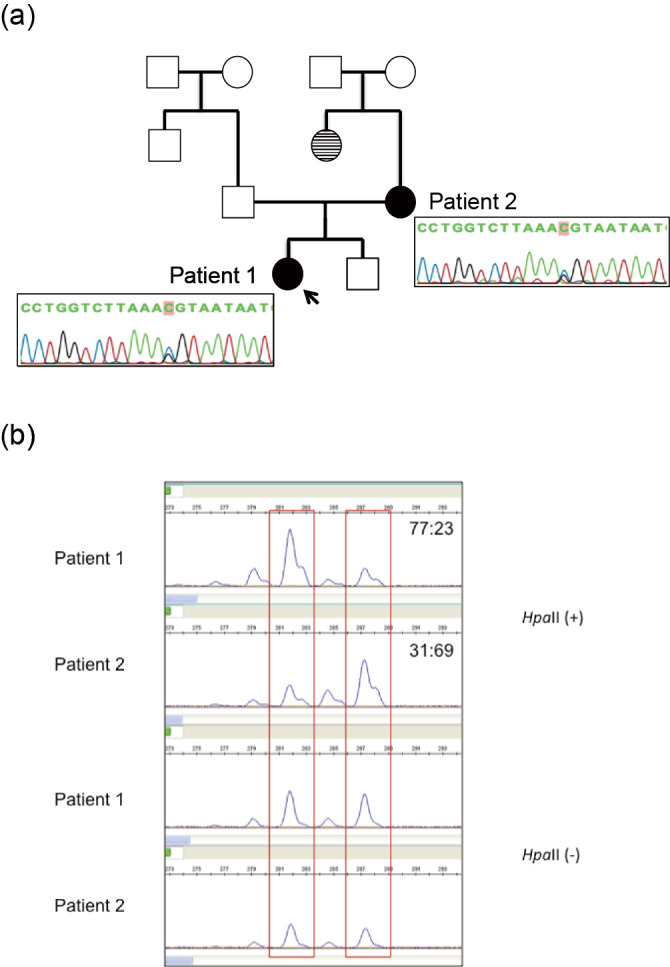
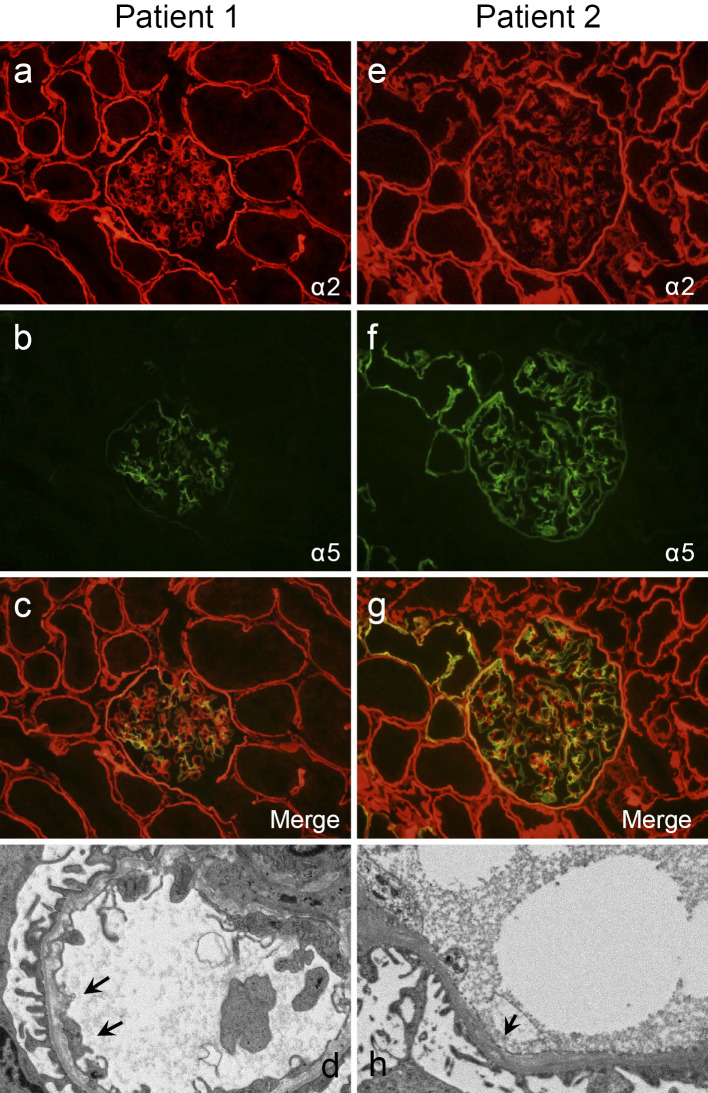
We herein report a novel mutation in a Japanese family with an X-linked Alport syndrome (AS) mutation in COL4A5. Patient 1 was a 2-year-old Japanese girl. She and her mother (patient 2) had a history of proteinuria and hematuria without renal dysfunction, deafness, or ocular abnormalities. Pathological findings were consistent with AS, and a genetic analysis revealed that both patients had a heterozygous mutation (c.2767G>C) in exon 32. In summary, the identification of mutations and characteristic pathological findings was useful in making a diagnosis of AS. For a close long-term follow-up, the early detection and treatment of women with X-linked AS are important.
我们在此报告一个日本家庭中COL4A5基因存在X连锁遗传性Alport综合征(AS)突变的新病例。患者1是一名2岁的日本女孩。她和她的母亲(患者2)有蛋白尿和血尿病史,但无肾功能障碍、耳聋或眼部异常。病理检查结果与AS一致,基因分析显示两名患者在第32外显子均有杂合突变(c.2767G>C)。总之,突变的鉴定和特征性病理检查结果有助于AS的诊断。对于长期密切随访而言,X连锁AS女性患者的早期检测和治疗很重要。