Timme Ruth E, Pettengill James B, Allard Marc W, Strain Errol, Barrangou Rodolphe, Wehnes Chris, Van Kessel Joann S, Karns Jeffrey S, Musser Steven M, Brown Eric W
Center for Food Safety and Applied Nutrition, U.S. Food and Drug Administration, College Park, MD.
Genome Biol Evol. 2013;5(11):2109-23. doi: 10.1093/gbe/evt159.
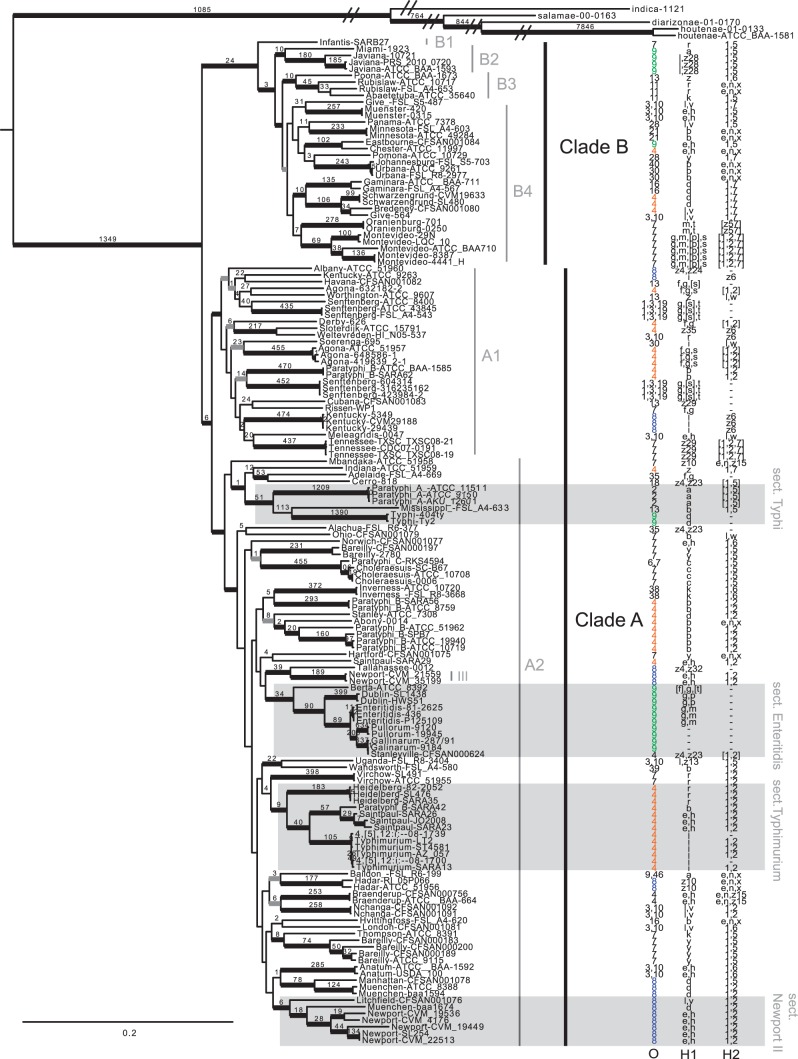
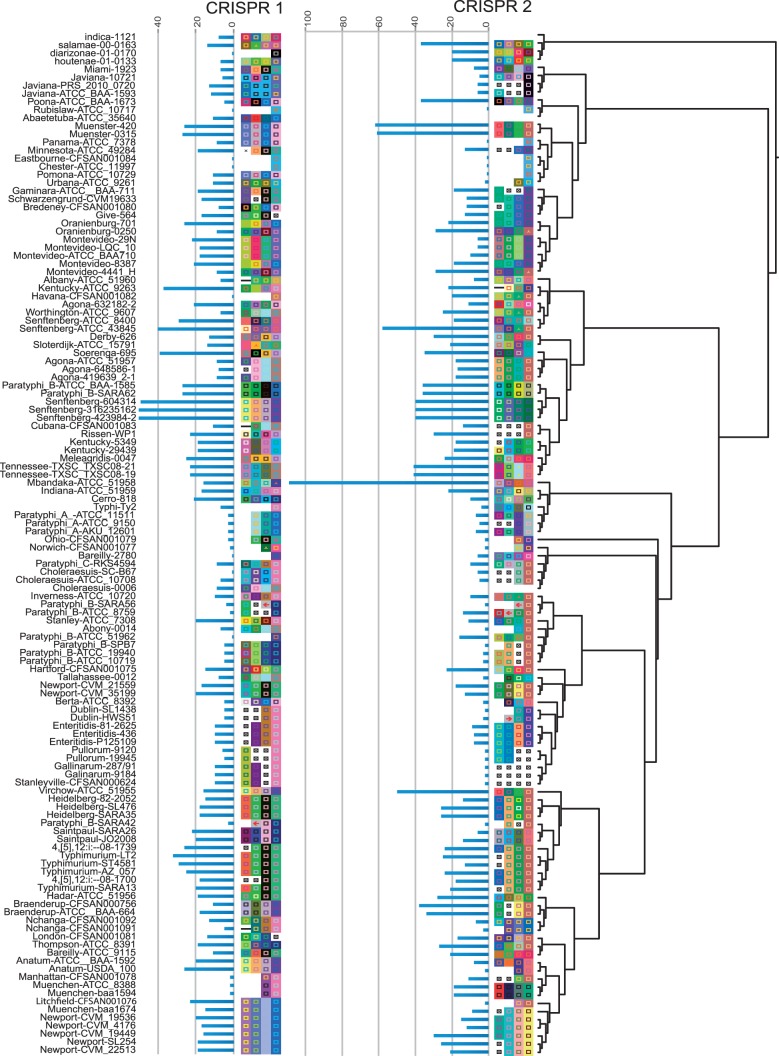
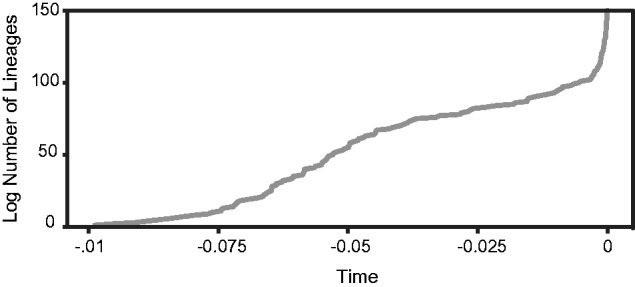
The enteric pathogen Salmonella enterica is one of the leading causes of foodborne illness in the world. The species is extremely diverse, containing more than 2,500 named serovars that are designated for their unique antigen characters and pathogenicity profiles-some are known to be virulent pathogens, while others are not. Questions regarding the evolution of pathogenicity, significance of antigen characters, diversity of clustered regularly interspaced short palindromic repeat (CRISPR) loci, among others, will remain elusive until a strong evolutionary framework is established. We present the first large-scale S. enterica subsp. enterica phylogeny inferred from a new reference-free k-mer approach of gathering single nucleotide polymorphisms (SNPs) from whole genomes. The phylogeny of 156 isolates representing 78 serovars (102 were newly sequenced) reveals two major lineages, each with many strongly supported sublineages. One of these lineages is the S. Typhi group; well nested within the phylogeny. Lineage-through-time analyses suggest there have been two instances of accelerated rates of diversification within the subspecies. We also found that antigen characters and CRISPR loci reveal different evolutionary patterns than that of the phylogeny, suggesting that a horizontal gene transfer or possibly a shared environmental acquisition might have influenced the present character distribution. Our study also shows the ability to extract reference-free SNPs from a large set of genomes and then to use these SNPs for phylogenetic reconstruction. This automated, annotation-free approach is an important step forward for bacterial disease tracking and in efficiently elucidating the evolutionary history of highly clonal organisms.
肠道病原体肠炎沙门氏菌是全球食源性疾病的主要病因之一。该菌种极其多样,包含2500多个命名血清型,这些血清型根据其独特的抗原特征和致病性概况来命名——有些是已知的强毒病原体,而有些则不是。在建立强大的进化框架之前,关于致病性的演变、抗原特征的意义、成簇规律间隔短回文重复序列(CRISPR)位点的多样性等问题仍将难以捉摸。我们展示了首个大规模肠炎沙门氏菌亚种肠炎沙门氏菌的系统发育树,该系统发育树是通过一种新的无需参考基因组的k-mer方法从全基因组中收集单核苷酸多态性(SNP)推断而来的。代表78个血清型的156株菌株(102株为新测序菌株)的系统发育树揭示了两个主要谱系,每个谱系都有许多得到有力支持的亚谱系。其中一个谱系是伤寒沙门氏菌组,很好地嵌套在系统发育树中。谱系随时间的分析表明,该亚种内有两次多样化加速的情况。我们还发现,抗原特征和CRISPR位点揭示了与系统发育树不同的进化模式,这表明水平基因转移或可能的共同环境获得可能影响了当前的特征分布。我们的研究还展示了从大量基因组中提取无需参考基因组的SNP,然后将这些SNP用于系统发育重建的能力。这种自动化、无注释的方法是细菌疾病追踪以及有效阐明高度克隆生物进化历史方面向前迈出的重要一步。