López de Maturana Rakel, Lang Valérie, Zubiarrain Amaia, Sousa Amaya, Vázquez Nerea, Gorostidi Ana, Águila Julio, López de Munain Adolfo, Rodríguez Manuel, Sánchez-Pernaute Rosario
Laboratory of Stem Cells and Neural Repair, Inbiomed, Paseo Mikeletegi, 81, E-20009, San Sebastian, Spain.
Laboratory of Ubiquitylation and Cancer Molecular Biology, Inbiomed, San Sebastian, Spain.
J Neuroinflammation. 2016 Nov 18;13(1):295. doi: 10.1186/s12974-016-0761-x.
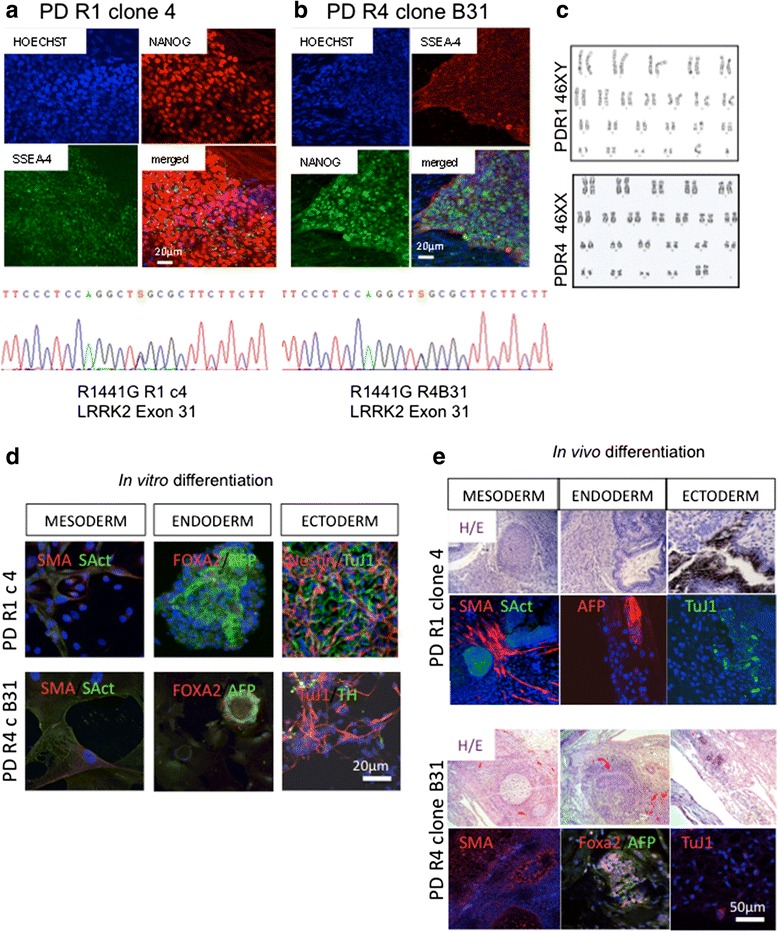
Mutations in leucine-rich repeat kinase 2 (LRRK2) contribute to both familial and idiopathic forms of Parkinson's disease (PD). Neuroinflammation is a key event in neurodegeneration and aging, and there is mounting evidence of LRRK2 involvement in inflammatory pathways. In a previous study, we described an alteration of the inflammatory response in dermal fibroblasts from PD patients expressing the G2019S and R1441G mutations in LRRK2.
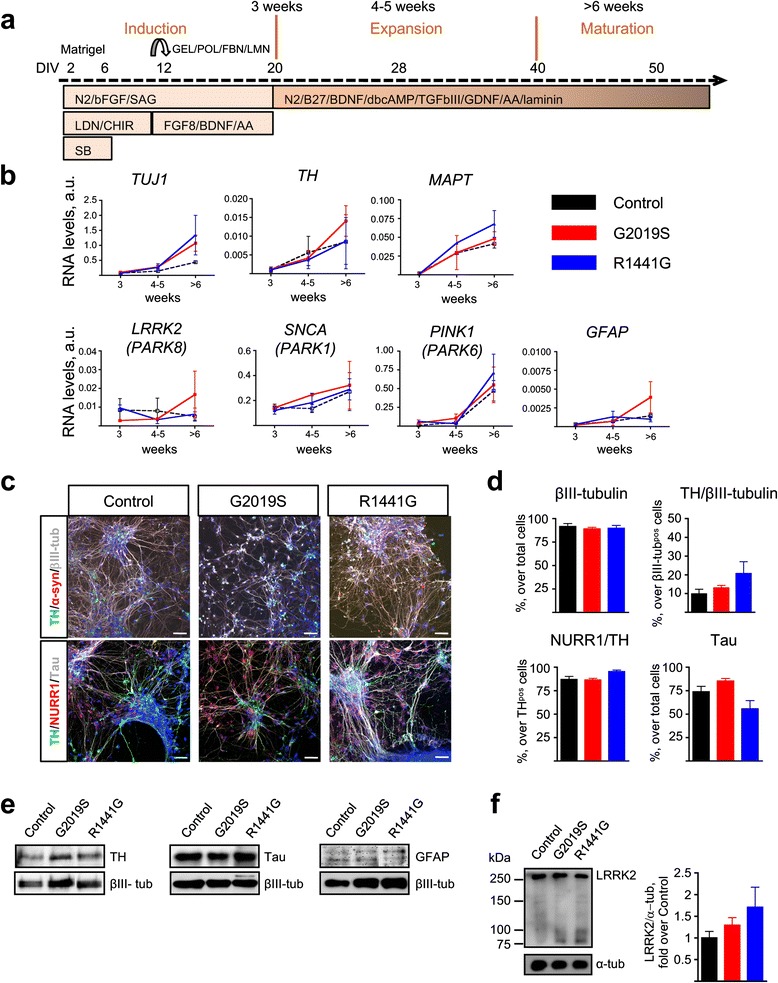
Taking advantage of cellular reprogramming, we generated induced pluripotent stem cell (iPSC) lines and neurons thereafter, harboring LRRK2 and LRRK2 mutations. We used gene silencing and functional reporter assays to characterize the effect of the mutations. We examined the temporal profile of TNFα-induced changes in proteins of the NF-κB pathway and optimized western blot analysis to capture α-synuclein dynamics. The effects of the mutations and interventions were analyzed by two-way ANOVA tests with respect to corresponding controls.
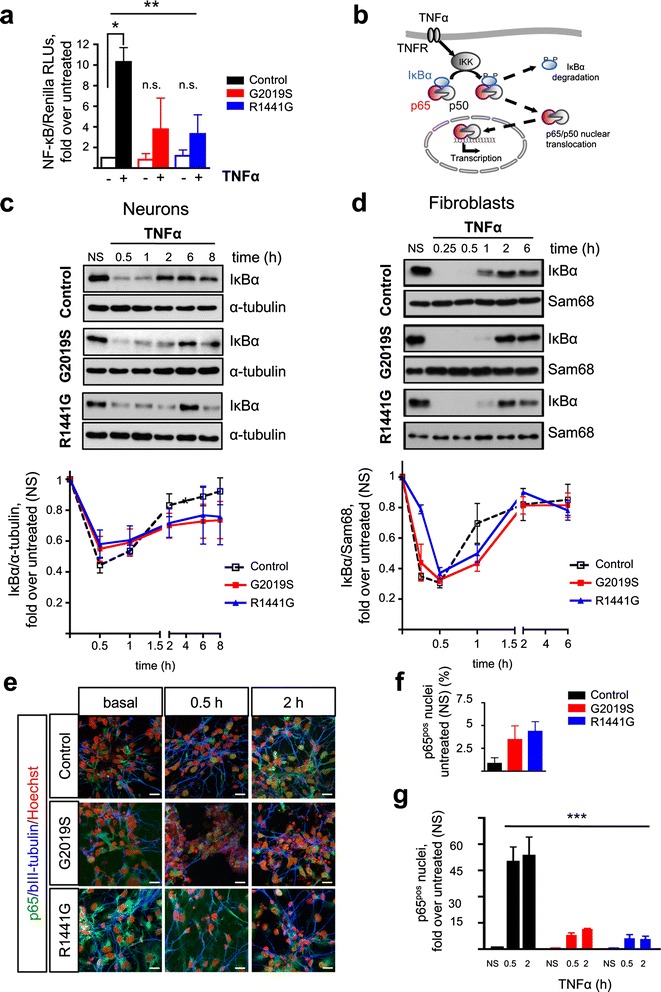
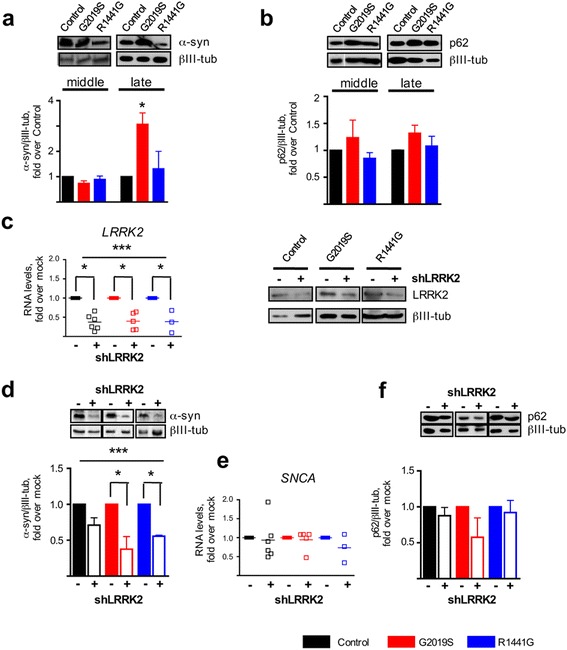
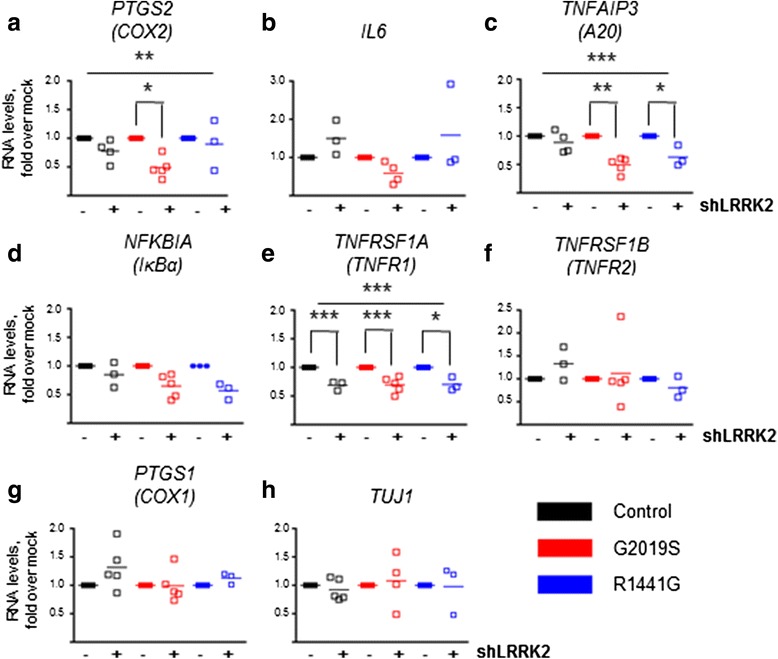
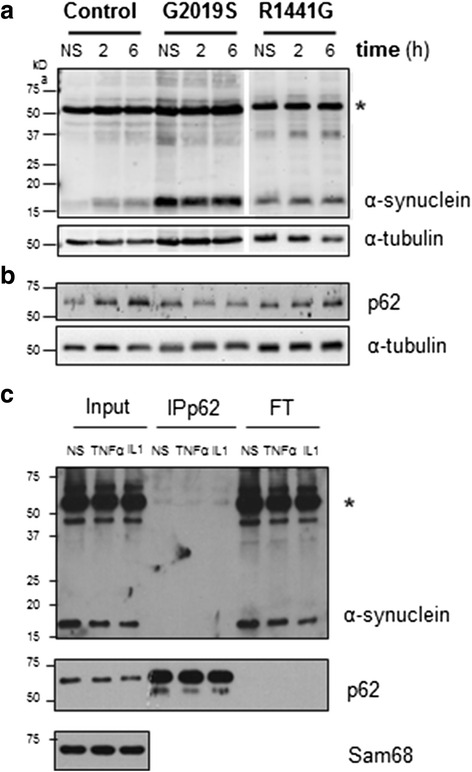
LRRK2 silencing decreased α-synuclein protein levels in mutated neurons and modified NF-κB transcriptional targets, such as PTGS2 (COX-2) and TNFAIP3 (A20). We next tested whether NF-κB and α-synuclein pathways converged and found that TNFα modulated α-synuclein levels, although we could not detect an effect of LRRK2 mutations, partly because of the individual variability. Nevertheless, we confirmed NF-κB dysregulation in mutated neurons, as shown by a protracted recovery of IκBα and a clear impairment in p65 nuclear translocation in the LRRK2 mutants.
Altogether, our results show that LRRK2 mutations affect α-synuclein regulation and impair NF-κB canonical signaling in iPSC-derived neurons. TNFα modulated α-synuclein proteostasis but was not modified by the LRRK2 mutations in this paradigm. These results strengthen the link between LRRK2 and the innate immunity system underscoring the involvement of inflammatory pathways in the neurodegenerative process in PD.
富含亮氨酸重复激酶2(LRRK2)的突变导致帕金森病(PD)的家族性和特发性形式。神经炎症是神经退行性变和衰老中的关键事件,越来越多的证据表明LRRK2参与炎症途径。在先前的一项研究中,我们描述了来自表达LRRK2中G2019S和R1441G突变的PD患者的真皮成纤维细胞中炎症反应的改变。
利用细胞重编程技术,我们随后生成了携带LRRK2和LRRK2突变的诱导多能干细胞(iPSC)系和神经元。我们使用基因沉默和功能报告分析来表征突变的影响。我们检查了TNFα诱导的NF-κB途径蛋白变化的时间概况,并优化了蛋白质印迹分析以捕获α-突触核蛋白的动态变化。通过双向方差分析测试分析突变和干预相对于相应对照的影响。
LRRK2沉默降低了突变神经元中α-突触核蛋白的水平,并改变了NF-κB转录靶点,如PTGS2(COX-2)和TNFAIP3(A20)。接下来,我们测试了NF-κB和α-突触核蛋白途径是否汇聚,发现TNFα调节α-突触核蛋白水平,尽管我们未检测到LRRK2突变的影响,部分原因是个体变异性。然而,我们证实了突变神经元中NF-κB失调,如LRRK2突变体中IκBα的持久恢复和p65核转位的明显受损所示。
总之,我们的结果表明LRRK2突变影响α-突触核蛋白的调节,并损害iPSC衍生神经元中的NF-κB经典信号传导。TNFα调节α-突触核蛋白的蛋白质稳态,但在该范例中未被LRRK2突变所改变。这些结果加强了LRRK2与先天免疫系统之间的联系,强调了炎症途径在PD神经退行性过程中的参与。