Lekli Istvan, Haines David Donald, Balla Gyorgy, Tosaki Arpad
Department of Pharmacology, Faculty of Pharmacy, University of Debrecen, Debrecen, Hungary.
Department of Pediatrics, Medical and Health Science Center, University of Debrecen, Debrecen, Hungary.
J Cell Mol Med. 2017 Jun;21(6):1058-1072. doi: 10.1111/jcmm.13053. Epub 2016 Dec 20.
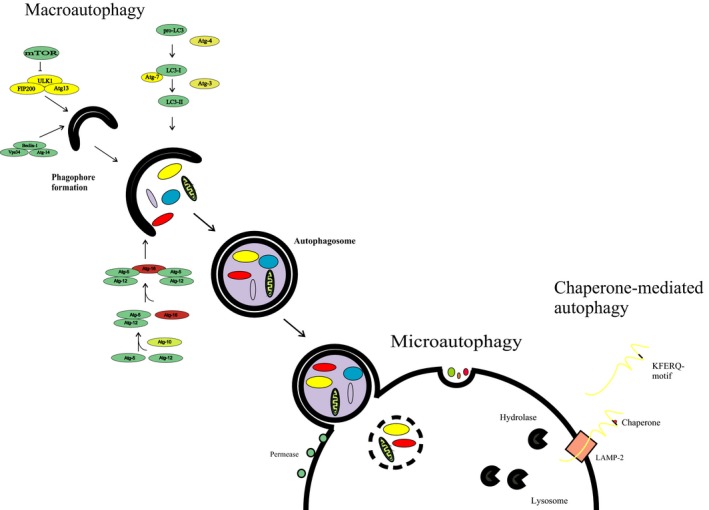
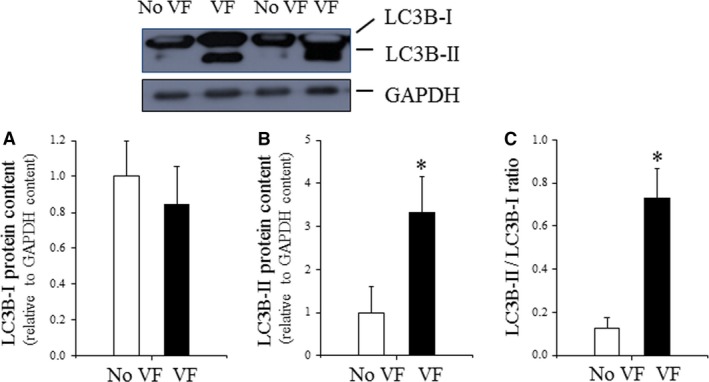
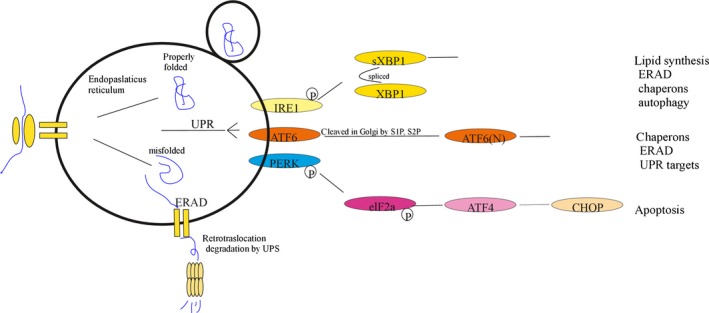
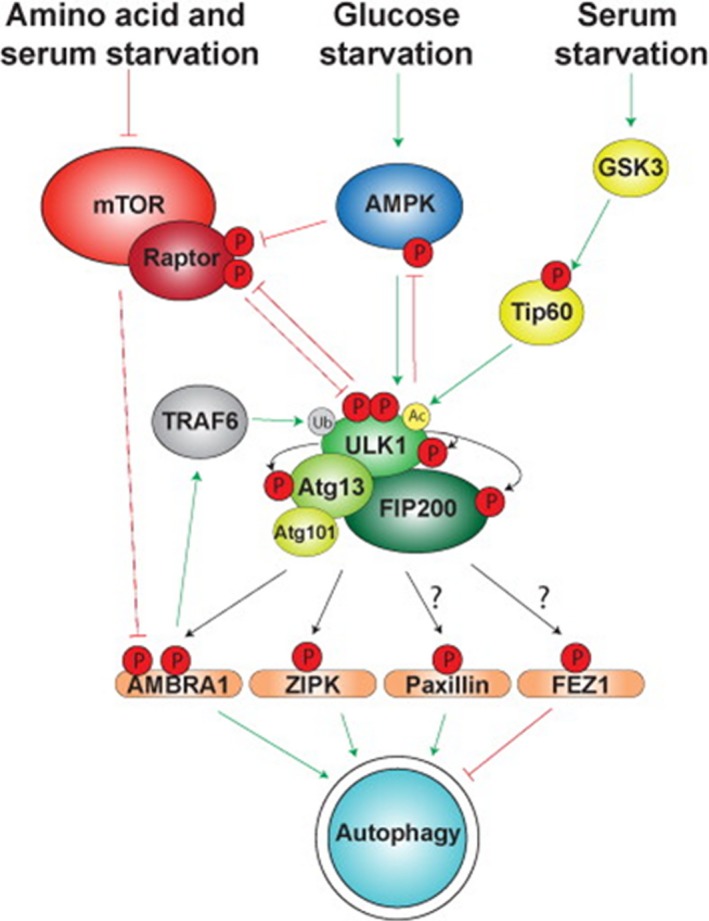
Oxidative stress placed on tissues that involved in pathogenesis of a disease activates compensatory metabolic changes, such as DNA damage repair that in turn causes intracellular accumulation of detritus and 'proteotoxic stress', leading to emergence of 'senescent' cellular phenotypes, which express high levels of inflammatory mediators, resulting in degradation of tissue function. Proteotoxic stress resulting from hyperactive inflammation following reperfusion of ischaemic tissue causes accumulation of proteinaceous debris in cells of the heart in ways that cause potentially fatal arrhythmias, in particular ventricular fibrillation (VF). An adaptive response to VF is occurrence of autophagy, an intracellular bulk degradation of damaged macromolecules and organelles that may restore cellular and tissue homoeostasis, improving chances for recovery. Nevertheless, depending on the type and intensity of stressors and inflammatory responses, autophagy may become pathological, resulting in excessive cell death. The present review examines the multilayered defences that cells have evolved to reduce proteotoxic stress by degradation of potentially toxic material beginning with endoplasmic reticulum-associated degradation, and the unfolded protein response, which are mechanisms for removal from the endoplasmic reticulum of misfolded proteins, and then progressing through the stages of autophagy, including descriptions of autophagosomes and related vesicular structures which process material for degradation and autophagy-associated proteins including Beclin-1 and regulatory complexes. The physiological roles of each mode of proteotoxic defence will be examined along with consideration of how emerging understanding of autophagy, along with a newly discovered regulatory cell type called telocytes, may be used to augment existing strategies for the prevention and management of cardiovascular disease.
疾病发病机制中涉及的组织所承受的氧化应激会激活代偿性代谢变化,比如DNA损伤修复,这反过来会导致细胞内碎屑积累和“蛋白毒性应激”,进而导致“衰老”细胞表型的出现,这些细胞会表达高水平的炎症介质,从而导致组织功能退化。缺血组织再灌注后炎症反应过度活跃所导致的蛋白毒性应激,会使心脏细胞中蛋白质碎片以可能引发致命性心律失常(尤其是心室颤动,VF)的方式积累。对VF的一种适应性反应是自噬的发生,自噬是细胞内对受损大分子和细胞器进行大规模降解的过程,可能会恢复细胞和组织的稳态,提高恢复的几率。然而,根据应激源和炎症反应的类型及强度,自噬可能会变得病理性,导致过度的细胞死亡。本综述探讨了细胞进化出的多层防御机制,这些机制通过降解潜在有毒物质来减少蛋白毒性应激,首先是内质网相关降解和未折叠蛋白反应,它们是从内质网中清除错误折叠蛋白质的机制,然后进入自噬阶段,包括对自噬体及相关囊泡结构的描述,这些结构处理用于降解的物质以及自噬相关蛋白,包括Beclin-1和调节复合物。每种蛋白毒性防御模式的生理作用将得到探讨,同时还会考虑对自噬的新认识,以及一种新发现的称为间充质干细胞的调节细胞类型,如何可用于增强现有的心血管疾病预防和管理策略。