Song Qiaoling, Sun Xiaoxiao, Guo Hui, Yu Qiang
Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 201203 Shanghai, China.
University of Chinese Academy of Sciences, 100049 Beijing, China.
Oncotarget. 2017 Jan 17;8(3):5003-5015. doi: 10.18632/oncotarget.14009.
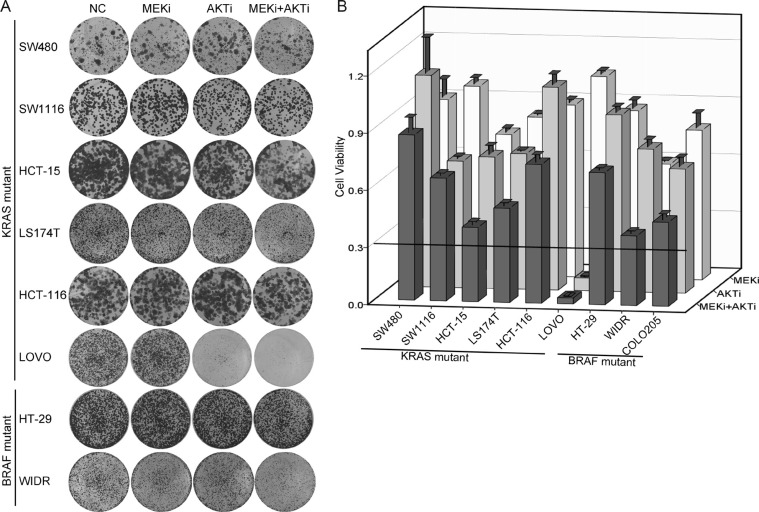
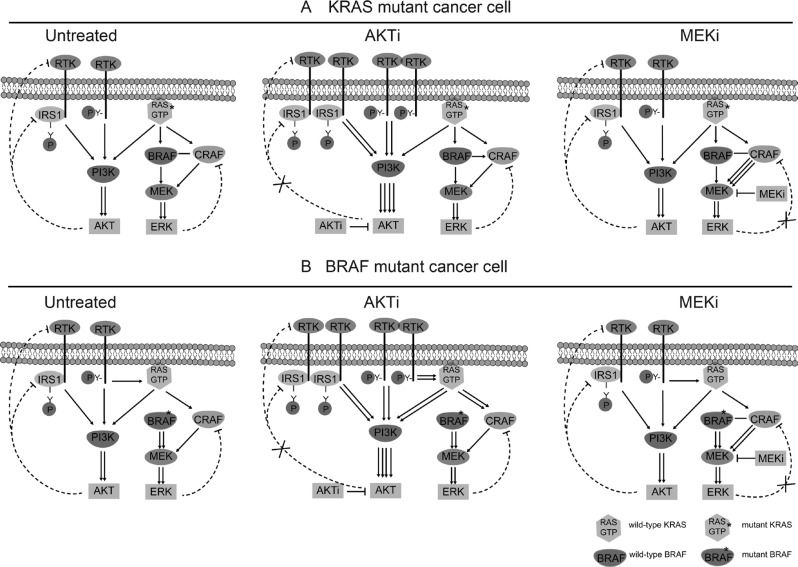
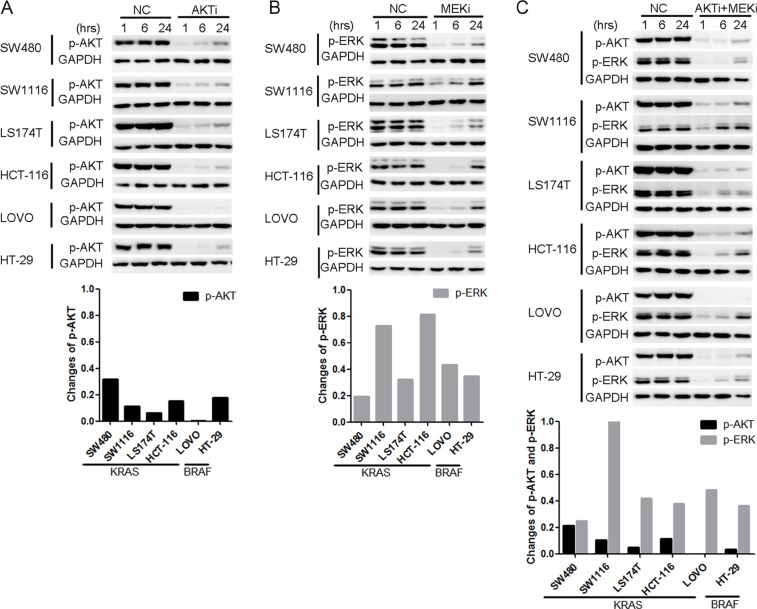
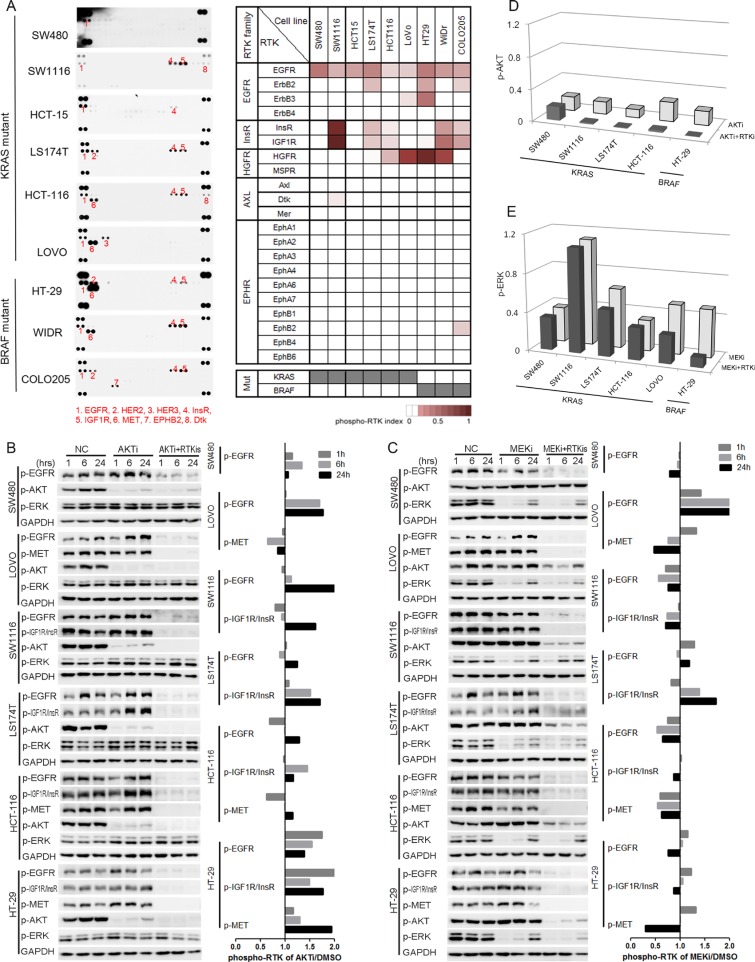
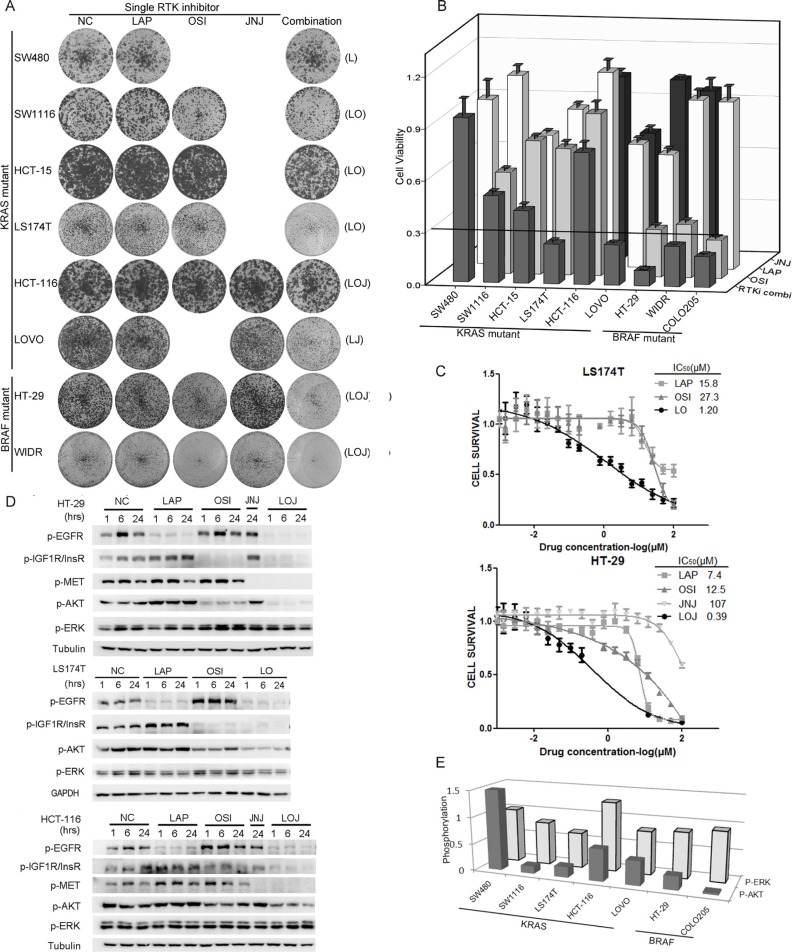
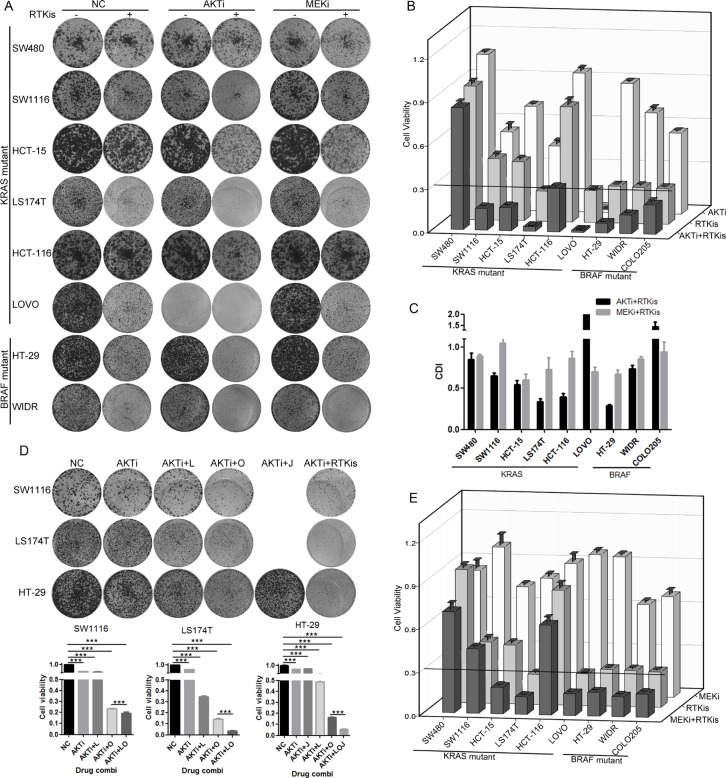
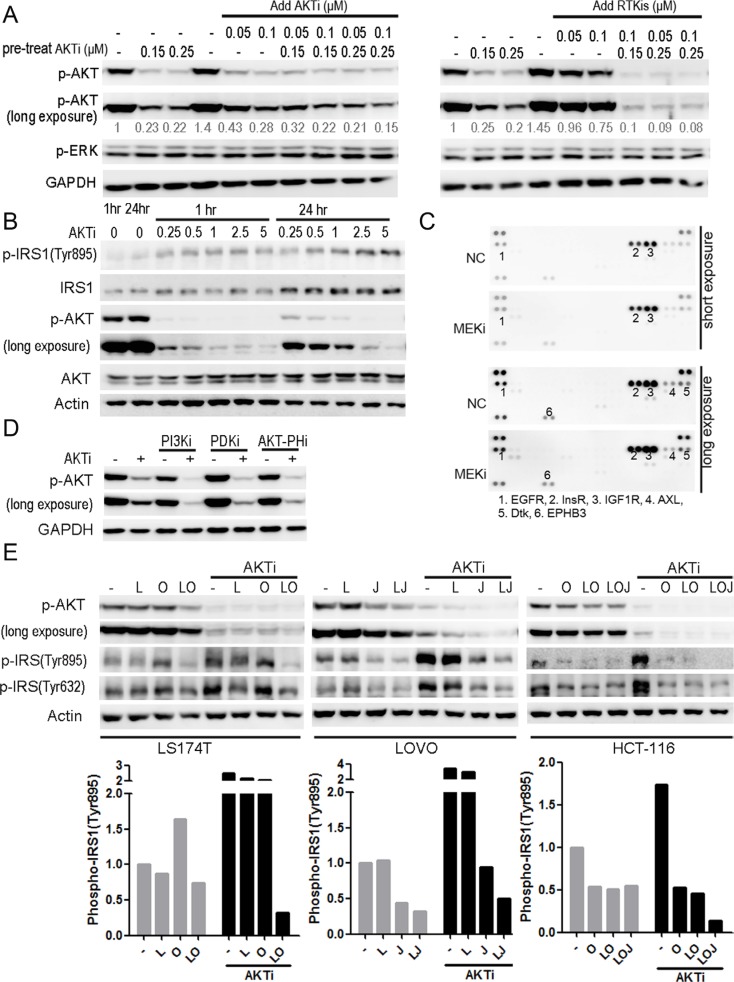
Receptor tyrosine kinase (RTK) signaling pathways are frequently activated in cancer cells due to mutations of RTKs and/or their downstream signaling proteins such as KRAS and BRAF. About 40% colorectal cancers (CRCs) contain KRAS or BRAF mutant genes and are resistant to treatments with individual inhibitors of RTKs, AKT, MEK, or BRAF. Therefore, an understanding of the molecular mechanisms of the drug resistance is necessary for developing effective strategies to treat the diseases. Here we report the discovery of an AKT/ERK reactivation mechanism that account for the cancer cell resistance to the AKT and MEK inhibitors treatments. The reactivations of AKT and ERK after the AKT or MEK inhibitor treatment were caused by a relief of an AKT or ERK-mediated feedback inhibition of the RTKs and/or their downstream pathways. A combination of RTK inhibitors, based on the RTK activation/phosphorylation profile, synergized with the AKT inhibitor, but not the MEK inhibitor, to completely inhibit the AKT phosphorylation and to block the growth of KRAS/BRAF mutant CRC cells. These results underscored the importance of AKT and the AKT feedback signaling to cancer cell growth and offered a novel therapeutic approach for the treatment of KRAS/BRAF mutant CRC cells.
受体酪氨酸激酶(RTK)信号通路在癌细胞中经常被激活,这是由于RTK及其下游信号蛋白(如KRAS和BRAF)发生突变所致。约40%的结直肠癌(CRC)含有KRAS或BRAF突变基因,并且对RTK、AKT、MEK或BRAF的单独抑制剂治疗具有抗性。因此,了解耐药性的分子机制对于制定有效的疾病治疗策略是必要的。在此,我们报告了一种AKT/ERK重新激活机制的发现,该机制解释了癌细胞对AKT和MEK抑制剂治疗的抗性。AKT或MEK抑制剂治疗后AKT和ERK的重新激活是由RTK和/或其下游通路的AKT或ERK介导的反馈抑制的解除所引起的。基于RTK激活/磷酸化谱的RTK抑制剂组合与AKT抑制剂协同作用,但不与MEK抑制剂协同作用,以完全抑制AKT磷酸化并阻断KRAS/BRAF突变CRC细胞的生长。这些结果强调了AKT和AKT反馈信号对癌细胞生长的重要性,并为治疗KRAS/BRAF突变CRC细胞提供了一种新的治疗方法。