Neurometabolic Diseases Laboratory, Institut de Neuropatologia de Bellvitge, IDIBELL, Gran Via, 199, L'Hospitalet de Llobregat, 08908, Barcelona, Spain.
CIBERER U759, Center for Biomedical Research on Rare Diseases, Barcelona, Spain.
Acta Neuropathol. 2017 Feb;133(2):283-301. doi: 10.1007/s00401-016-1655-9. Epub 2016 Dec 21.
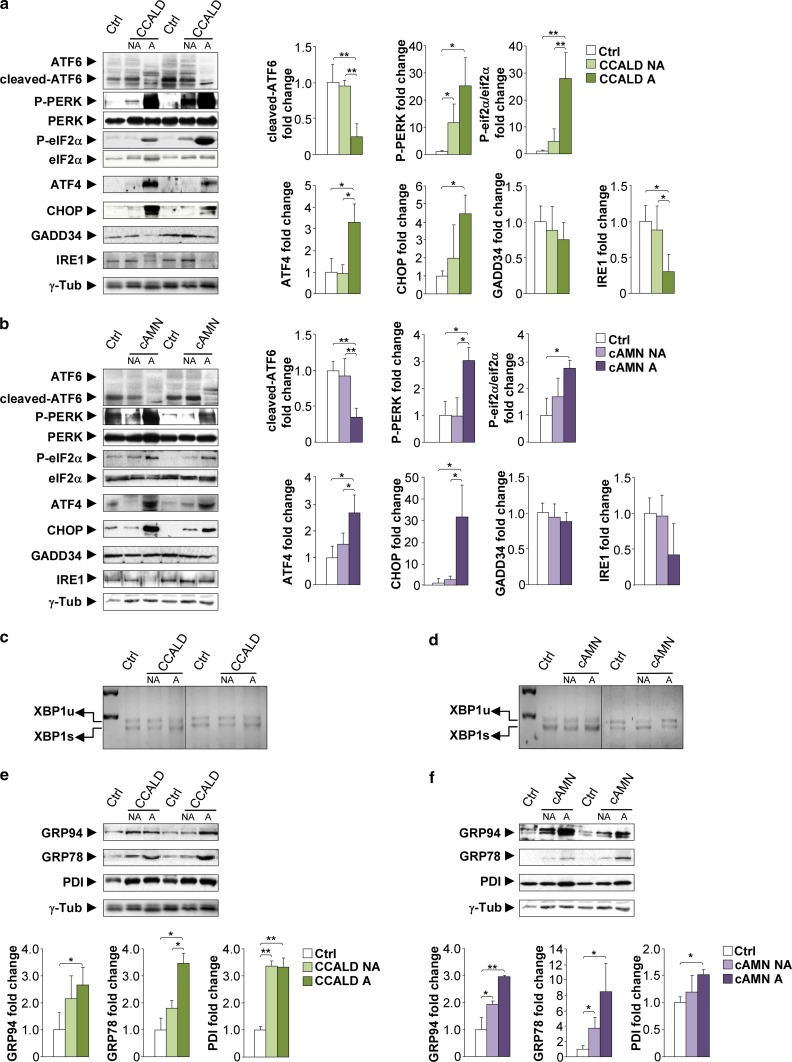
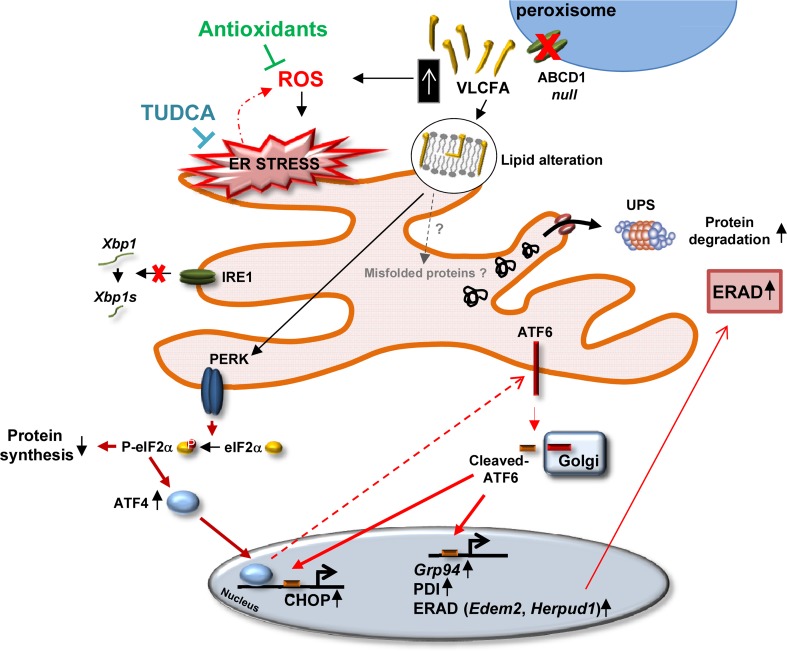
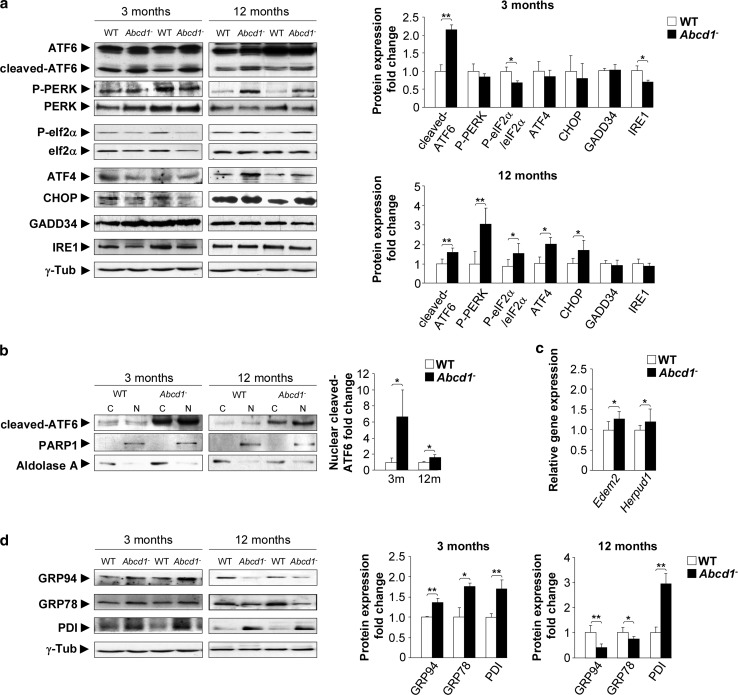
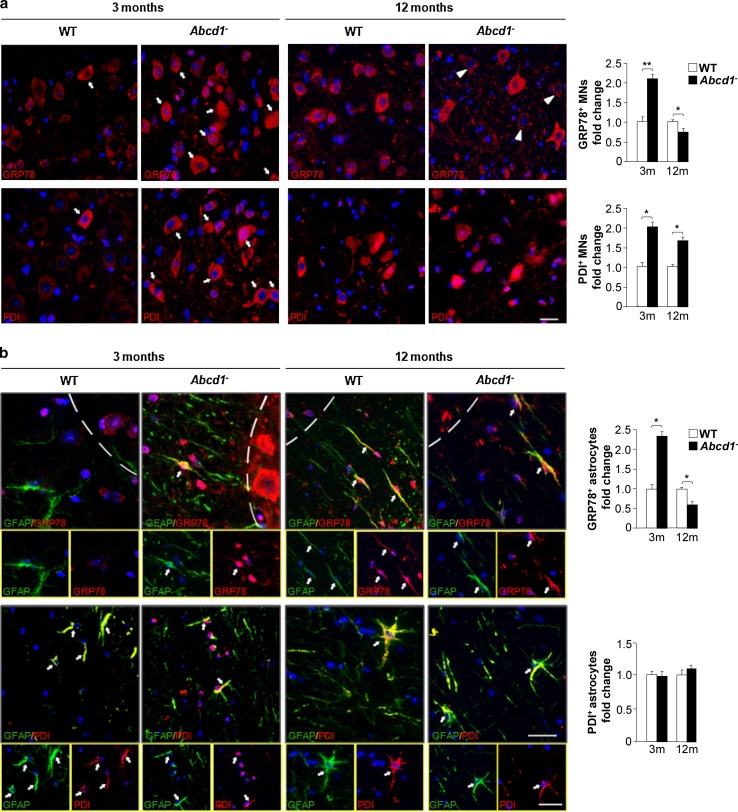
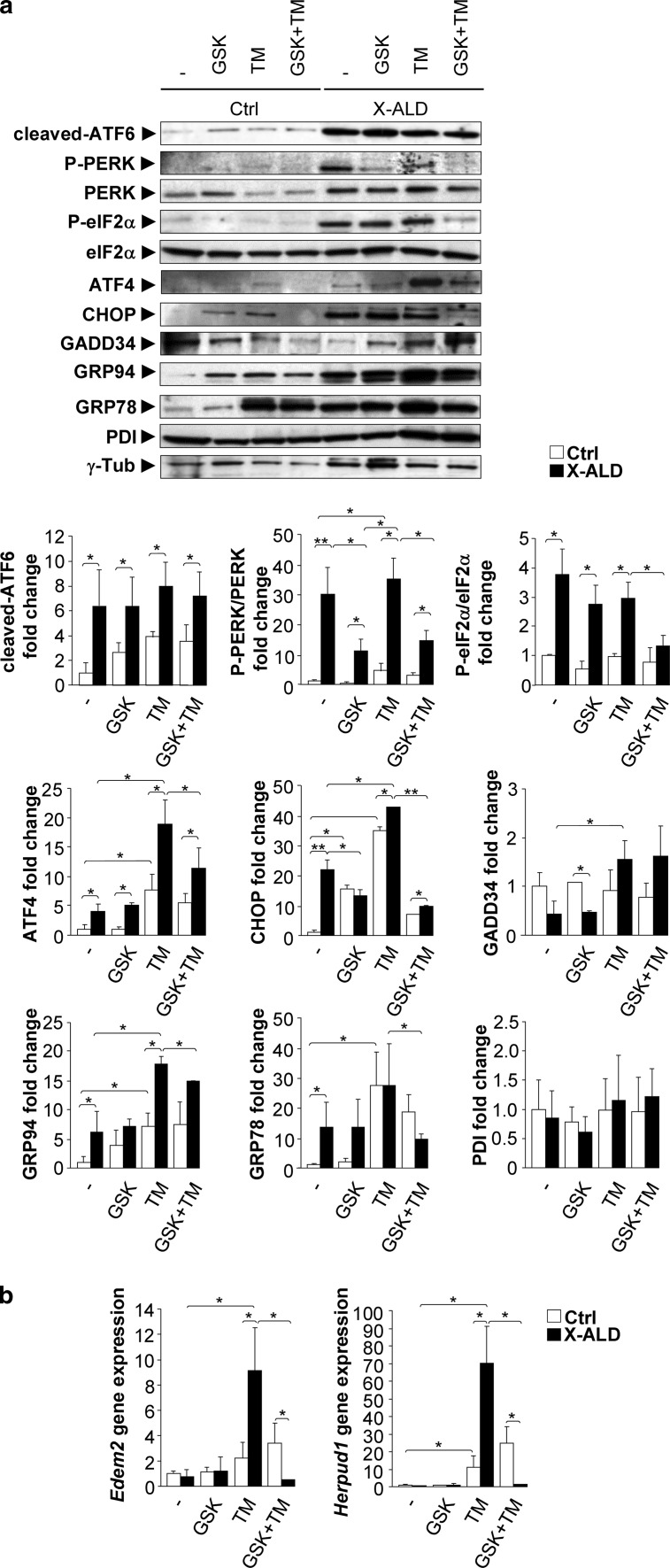
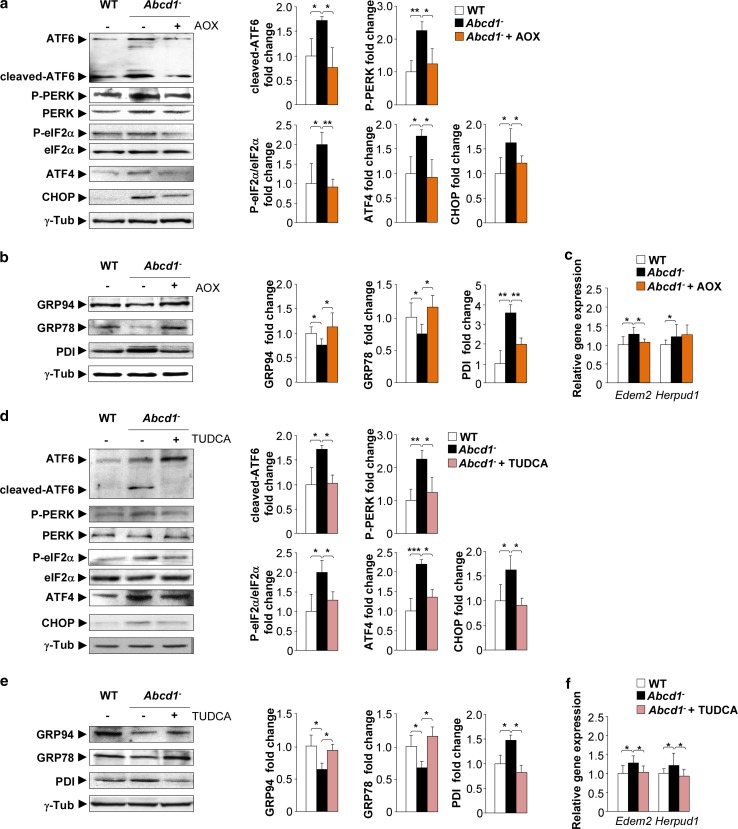
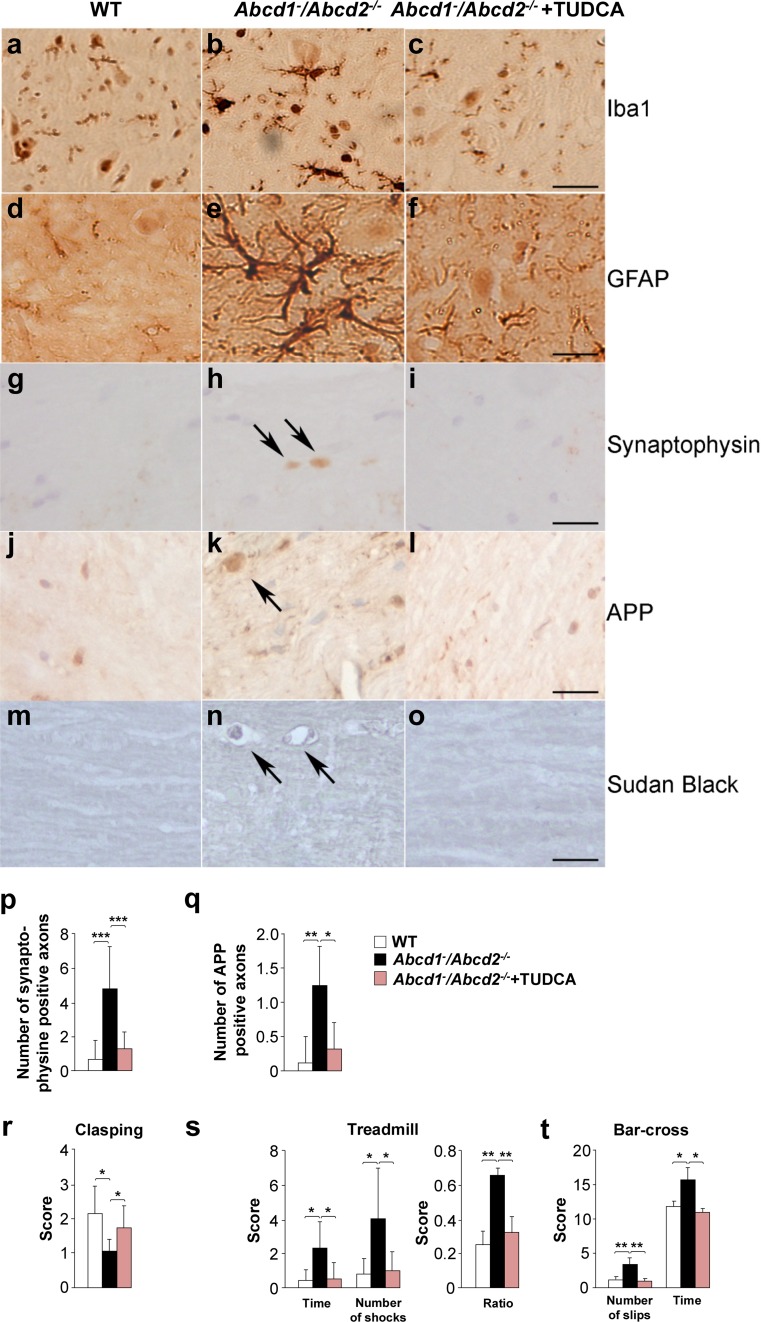
The activation of the highly conserved unfolded protein response (UPR) is prominent in the pathogenesis of the most prevalent neurodegenerative disorders, such as Alzheimer's disease (AD), Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS), which are classically characterized by an accumulation of aggregated or misfolded proteins. This activation is orchestrated by three endoplasmic reticulum (ER) stress sensors: PERK, ATF6 and IRE1. These sensors transduce signals that induce the expression of the UPR gene programme. Here, we first identified an early activator of the UPR and investigated the role of a chronically activated UPR in the pathogenesis of X-linked adrenoleukodystrophy (X-ALD), a neurometabolic disorder that is caused by ABCD1 malfunction; ABCD1 transports very long-chain fatty acids (VLCFA) into peroxisomes. The disease manifests as inflammatory demyelination in the brain or and/or degeneration of corticospinal tracts, thereby resulting in spastic paraplegia, with the accumulation of intracellular VLCFA instead of protein aggregates. Using X-ALD mouse model (Abcd1 and Abcd1 /Abcd2 mice) and X-ALD patient's fibroblasts and brain samples, we discovered an early engagement of the UPR. The response was characterized by the activation of the PERK and ATF6 pathways, but not the IRE1 pathway, showing a difference from the models of AD, PD or ALS. Inhibition of PERK leads to the disruption of homeostasis and increased apoptosis during ER stress induced in X-ALD fibroblasts. Redox imbalance appears to be the mechanism that initiates ER stress in X-ALD. Most importantly, we demonstrated that the bile acid tauroursodeoxycholate (TUDCA) abolishes UPR activation, which results in improvement of axonal degeneration and its associated locomotor impairment in Abcd1 /Abcd2 mice. Altogether, our preclinical data provide evidence for establishing the UPR as a key drug target in the pathogenesis cascade. Our study also highlights the potential role of TUDCA as a treatment for X-ALD and other axonopathies in which similar molecular mediators are implicated.
未折叠蛋白反应(UPR)的激活在最常见的神经退行性疾病的发病机制中很明显,如阿尔茨海默病(AD)、帕金森病(PD)和肌萎缩侧索硬化症(ALS),这些疾病的特征通常是聚集或错误折叠的蛋白质积累。这种激活是由三个内质网(ER)应激传感器协调的:PERK、ATF6 和 IRE1。这些传感器传递信号,诱导 UPR 基因程序的表达。在这里,我们首先鉴定了 UPR 的早期激活剂,并研究了慢性激活的 UPR 在 X 连锁肾上腺脑白质营养不良(X-ALD)发病机制中的作用,X-ALD 是一种由 ABCD1 功能障碍引起的神经代谢疾病;ABCD1 将长链脂肪酸(VLCFA)转运到过氧化物酶体中。该疾病表现为大脑中的炎症性脱髓鞘或和/或皮质脊髓束的变性,从而导致痉挛性截瘫,细胞内 VLCFA 的积累而不是蛋白质聚集。使用 X-ALD 小鼠模型(Abcd1 和 Abcd1 / Abcd2 小鼠)和 X-ALD 患者的成纤维细胞和脑组织样本,我们发现 UPR 早期参与。该反应的特征是 PERK 和 ATF6 途径的激活,但 IRE1 途径没有激活,与 AD、PD 或 ALS 的模型不同。PERK 抑制导致内质网应激诱导的 X-ALD 成纤维细胞中平衡破坏和细胞凋亡增加。氧化还原失衡似乎是 X-ALD 中内质网应激的启动机制。最重要的是,我们证明了胆酸牛磺熊脱氧胆酸(TUDCA)可消除 UPR 的激活,从而改善 Abcd1 / Abcd2 小鼠的轴突变性及其相关运动障碍。总之,我们的临床前数据为将 UPR 确立为发病机制级联中的关键药物靶点提供了证据。我们的研究还强调了 TUDCA 作为治疗 X-ALD 和其他涉及类似分子介质的轴突病的潜在作用。