Vaughan Timothy G, Welch David, Drummond Alexei J, Biggs Patrick J, George Tessy, French Nigel P
Centre for Computational Evolution, The University of Auckland, 1010, New Zealand
Department of Computer Science, The University of Auckland, 1010, New Zealand.
Genetics. 2017 Feb;205(2):857-870. doi: 10.1534/genetics.116.193425. Epub 2016 Dec 22.
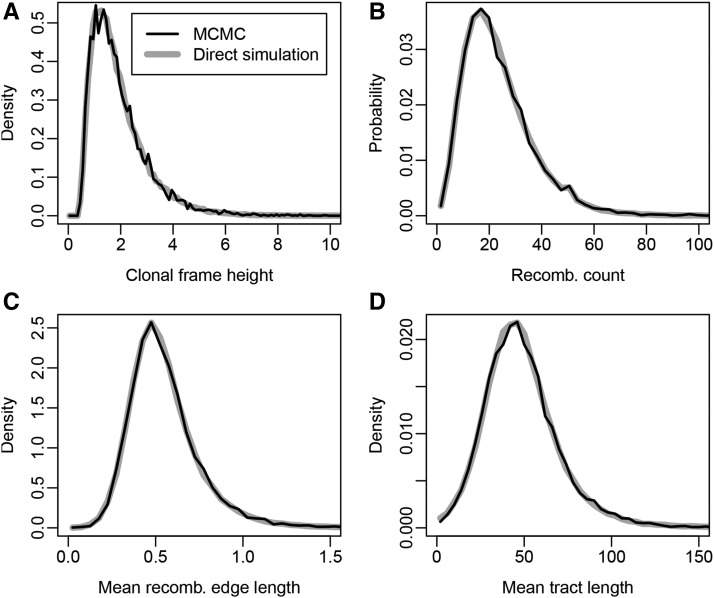
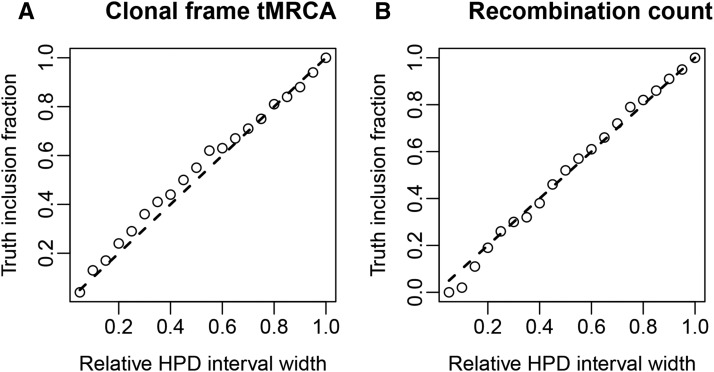
Homologous recombination is a central feature of bacterial evolution, yet it confounds traditional phylogenetic methods. While a number of methods specific to bacterial evolution have been developed, none of these permit joint inference of a bacterial recombination graph and associated parameters. In this article, we present a new method which addresses this shortcoming. Our method uses a novel Markov chain Monte Carlo algorithm to perform phylogenetic inference under the ClonalOrigin model. We demonstrate the utility of our method by applying it to ribosomal multilocus sequence typing data sequenced from pathogenic and nonpathogenic Escherichia coli serotype O157 and O26 isolates collected in rural New Zealand. The method is implemented as an open source BEAST 2 package, Bacter, which is available via the project web page at http://tgvaughan.github.io/bacter.
同源重组是细菌进化的一个核心特征,但它使传统的系统发育方法变得复杂。虽然已经开发了许多特定于细菌进化的方法,但这些方法都不允许联合推断细菌重组图谱和相关参数。在本文中,我们提出了一种新方法来解决这一缺点。我们的方法使用一种新颖的马尔可夫链蒙特卡罗算法,在克隆起源模型下进行系统发育推断。我们通过将其应用于从新西兰农村收集的致病性和非致病性大肠杆菌O157和O26血清型分离株测序得到的核糖体多位点序列分型数据,证明了我们方法的实用性。该方法作为一个开源的BEAST 2软件包Bacter实现,可通过项目网页http://tgvaughan.github.io/bacter获得。