Lazaris Charalampos, Kelly Stephen, Ntziachristos Panagiotis, Aifantis Iannis, Tsirigos Aristotelis
Department of Pathology, NYU School of Medicine, New York, NY, 10016, USA.
Laura and Isaac Perlmutter Cancer Center and Helen L. and Martin S. Kimmel Center for Stem Cell Biology, NYU School of Medicine, New York, NY, 10016, USA.
BMC Genomics. 2017 Jan 5;18(1):22. doi: 10.1186/s12864-016-3387-6.
Chromatin conformation capture techniques have evolved rapidly over the last few years and have provided new insights into genome organization at an unprecedented resolution. Analysis of Hi-C data is complex and computationally intensive involving multiple tasks and requiring robust quality assessment. This has led to the development of several tools and methods for processing Hi-C data. However, most of the existing tools do not cover all aspects of the analysis and only offer few quality assessment options. Additionally, availability of a multitude of tools makes scientists wonder how these tools and associated parameters can be optimally used, and how potential discrepancies can be interpreted and resolved. Most importantly, investigators need to be ensured that slight changes in parameters and/or methods do not affect the conclusions of their studies.
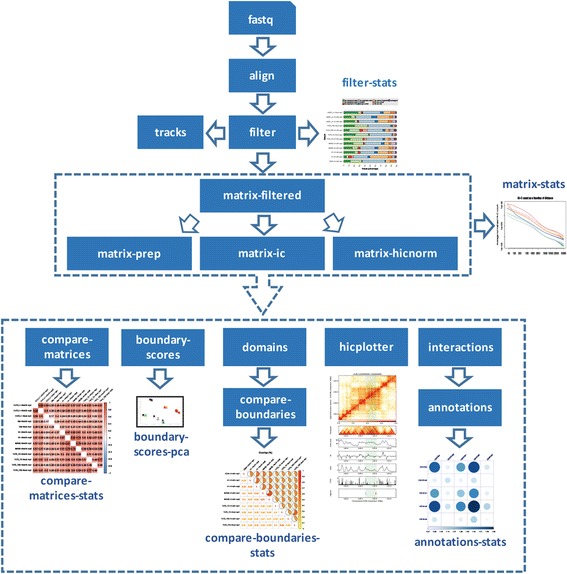
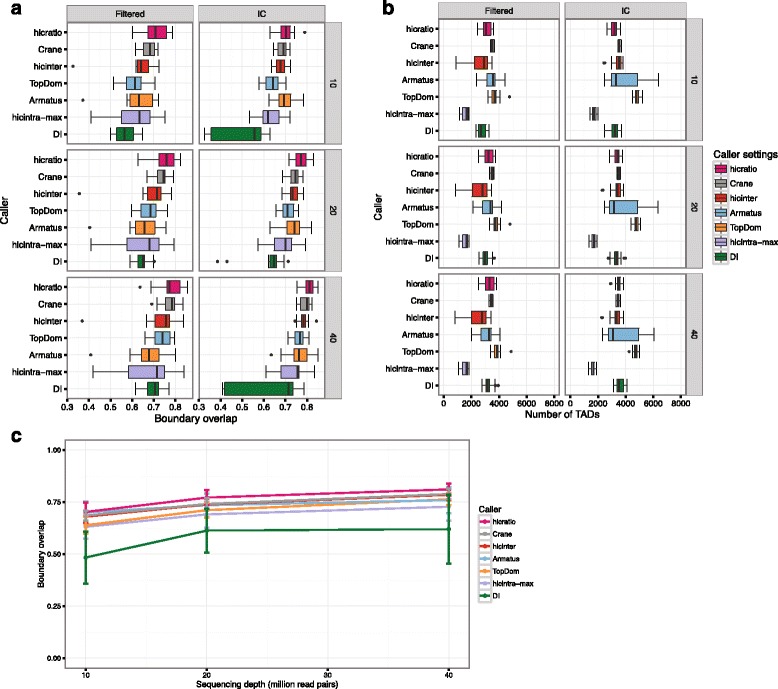
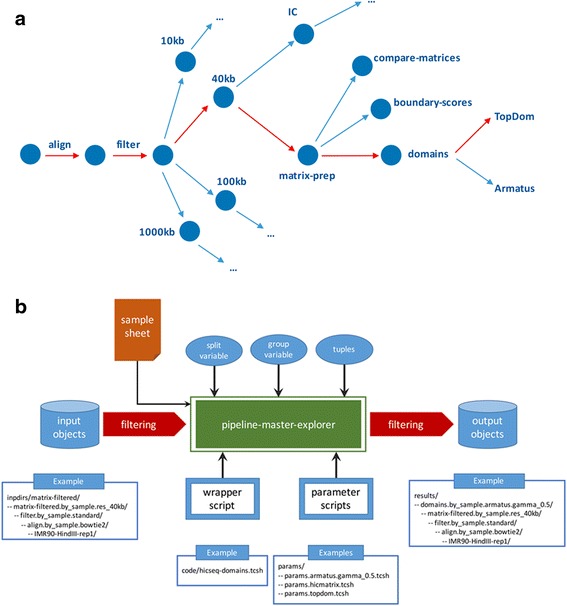
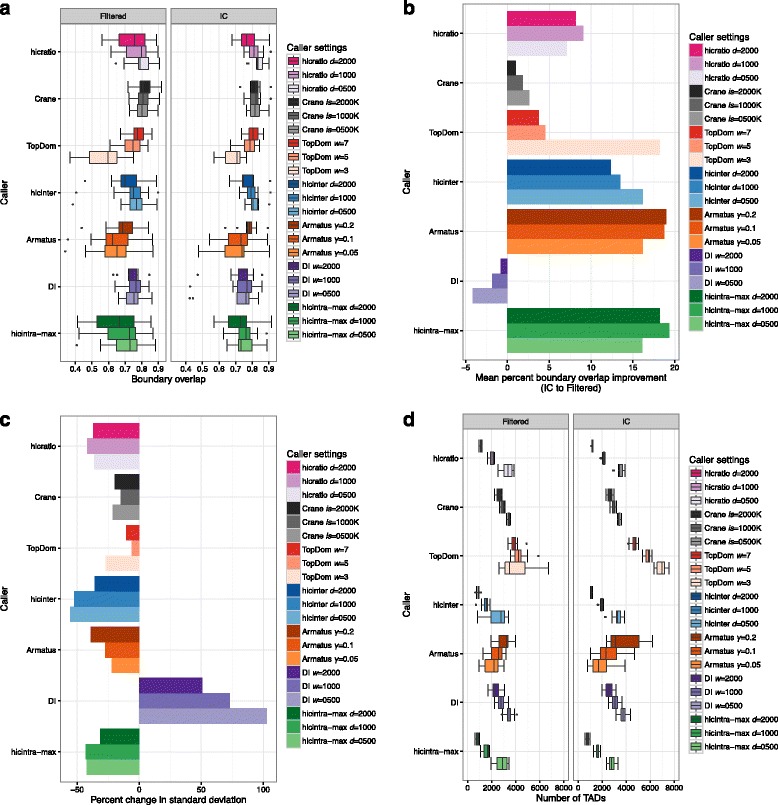
To address these issues (compare, explore and reproduce), we introduce HiC-bench, a configurable computational platform for comprehensive and reproducible analysis of Hi-C sequencing data. HiC-bench performs all common Hi-C analysis tasks, such as alignment, filtering, contact matrix generation and normalization, identification of topological domains, scoring and annotation of specific interactions using both published tools and our own. We have also embedded various tasks that perform quality assessment and visualization. HiC-bench is implemented as a data flow platform with an emphasis on analysis reproducibility. Additionally, the user can readily perform parameter exploration and comparison of different tools in a combinatorial manner that takes into account all desired parameter settings in each pipeline task. This unique feature facilitates the design and execution of complex benchmark studies that may involve combinations of multiple tool/parameter choices in each step of the analysis. To demonstrate the usefulness of our platform, we performed a comprehensive benchmark of existing and new TAD callers exploring different matrix correction methods, parameter settings and sequencing depths. Users can extend our pipeline by adding more tools as they become available.
HiC-bench consists an easy-to-use and extensible platform for comprehensive analysis of Hi-C datasets. We expect that it will facilitate current analyses and help scientists formulate and test new hypotheses in the field of three-dimensional genome organization.
在过去几年中,染色质构象捕获技术发展迅速,并以前所未有的分辨率为基因组组织提供了新的见解。Hi-C数据的分析复杂且计算量大,涉及多个任务,需要进行可靠的质量评估。这导致了几种用于处理Hi-C数据的工具和方法的开发。然而,大多数现有工具并未涵盖分析的所有方面,仅提供很少的质量评估选项。此外,众多工具的存在让科学家们疑惑如何能最佳地使用这些工具及其相关参数,以及如何解释和解决潜在的差异。最重要的是,研究人员需要确保参数和/或方法的微小变化不会影响其研究结论。
为了解决这些问题(比较、探索和重现),我们引入了HiC-bench,这是一个用于Hi-C测序数据全面且可重现分析的可配置计算平台。HiC-bench执行所有常见的Hi-C分析任务,例如比对、过滤、接触矩阵生成和标准化、拓扑结构域的识别、使用已发表的工具和我们自己的工具对特定相互作用进行评分和注释。我们还嵌入了各种执行质量评估和可视化的任务。HiC-bench被实现为一个强调分析可重复性的数据流平台。此外,用户可以以组合方式轻松地进行参数探索和不同工具的比较,同时考虑每个管道任务中的所有所需参数设置。这一独特功能有助于设计和执行复杂的基准研究,这些研究可能在分析的每个步骤中涉及多个工具/参数选择的组合。为了证明我们平台的实用性,我们对现有的和新的TAD调用工具进行了全面的基准测试,探索了不同的矩阵校正方法、参数设置和测序深度。用户可以在有更多可用工具时通过添加它们来扩展我们的管道。
HiC-bench是一个用于Hi-C数据集全面分析的易于使用且可扩展的平台。我们期望它将促进当前的分析,并帮助科学家们在三维基因组组织领域制定和测试新的假设。