Ahrenfeldt Johanne, Skaarup Carina, Hasman Henrik, Pedersen Anders Gorm, Aarestrup Frank Møller, Lund Ole
Center for Biological Sequence Analysis, DTU Bioinformatics, Technical University of Denmark, Kongens Lyngby, Denmark.
Department of Microbiology and Infection Control, Statens Serum Institute, Copenhagen, Denmark.
BMC Genomics. 2017 Jan 5;18(1):19. doi: 10.1186/s12864-016-3407-6.
Whole genome sequencing (WGS) is increasingly used in diagnostics and surveillance of infectious diseases. A major application for WGS is to use the data for identifying outbreak clusters, and there is therefore a need for methods that can accurately and efficiently infer phylogenies from sequencing reads. In the present study we describe a new dataset that we have created for the purpose of benchmarking such WGS-based methods for epidemiological data, and also present an analysis where we use the data to compare the performance of some current methods.
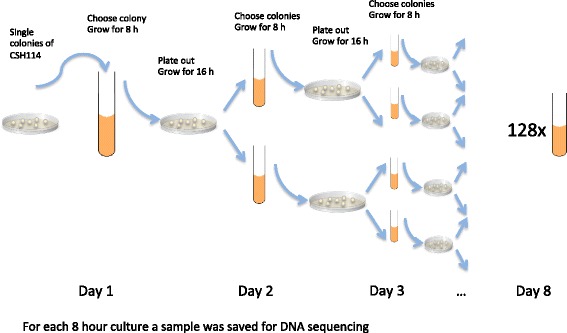
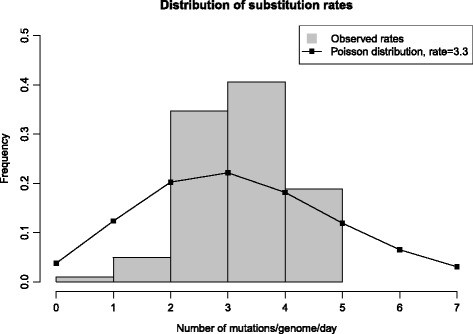
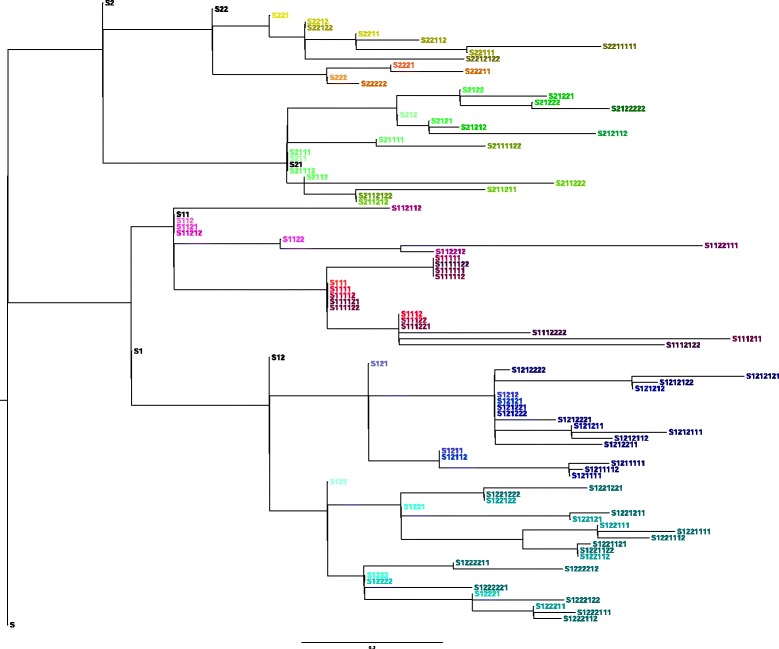
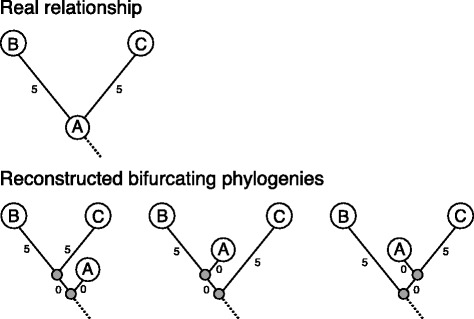
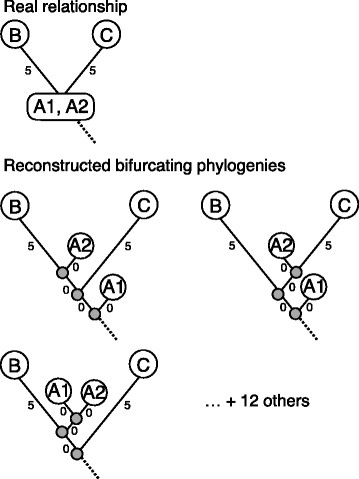
Our aim was to create a benchmark data set that mimics sequencing data of the sort that might be collected during an outbreak of an infectious disease. This was achieved by letting an E. coli hypermutator strain grow in the lab for 8 consecutive days, each day splitting the culture in two while also collecting samples for sequencing. The result is a data set consisting of 101 whole genome sequences with known phylogenetic relationship. Among the sequenced samples 51 correspond to internal nodes in the phylogeny because they are ancestral, while the remaining 50 correspond to leaves. We also used the newly created data set to compare three different online available methods that infer phylogenies from whole-genome sequencing reads: NDtree, CSI Phylogeny and REALPHY. One complication when comparing the output of these methods with the known phylogeny is that phylogenetic methods typically build trees where all observed sequences are placed as leafs, even though some of them are in fact ancestral. We therefore devised a method for post processing the inferred trees by collapsing short branches (thus relocating some leafs to internal nodes), and also present two new measures of tree similarity that takes into account the identity of both internal and leaf nodes.
Based on this analysis we find that, among the investigated methods, CSI Phylogeny had the best performance, correctly identifying 73% of all branches in the tree and 71% of all clades. We have made all data from this experiment (raw sequencing reads, consensus whole-genome sequences, as well as descriptions of the known phylogeny in a variety of formats) publicly available, with the hope that other groups may find this data useful for benchmarking and exploring the performance of epidemiological methods. All data is freely available at: https://cge.cbs.dtu.dk/services/evolution_data.php .
全基因组测序(WGS)在传染病的诊断和监测中应用越来越广泛。WGS的一个主要应用是利用数据识别疫情集群,因此需要能够从测序读数中准确、高效地推断系统发育的方法。在本研究中,我们描述了一个新创建的数据集,用于对基于WGS的流行病学数据方法进行基准测试,并展示了一项分析,其中我们使用该数据比较了一些现有方法的性能。
我们的目标是创建一个模拟传染病爆发期间可能收集的测序数据类型的基准数据集。这是通过让一株大肠杆菌高突变株在实验室连续培养8天来实现的,每天将培养物分成两份,同时收集样本进行测序。结果得到了一个由101个具有已知系统发育关系的全基因组序列组成的数据集。在测序样本中,51个对应于系统发育中的内部节点,因为它们是祖先序列,而其余50个对应于叶节点。我们还使用新创建的数据集比较了三种不同的在线可用方法,这些方法从全基因组测序读数中推断系统发育:NDtree、CSI Phylogeny和REALPHY。将这些方法的输出与已知系统发育进行比较时的一个复杂情况是,系统发育方法通常构建的树中所有观察到的序列都被放置为叶节点,即使其中一些实际上是祖先序列。因此,我们设计了一种通过合并短分支(从而将一些叶节点重新定位到内部节点)对推断树进行后处理的方法,并提出了两种新的树相似性度量,同时考虑了内部节点和叶节点的身份。
基于此分析,我们发现,在所研究的方法中,CSI Phylogeny性能最佳,正确识别了树中所有分支的73%和所有进化枝的71%。我们已将该实验的所有数据(原始测序读数、一致全基因组序列以及各种格式的已知系统发育描述)公开,希望其他研究团队会发现这些数据对基准测试和探索流行病学方法的性能有用。所有数据均可在以下网址免费获取:https://cge.cbs.dtu.dk/services/evolution_data.php 。