Department of Microbiology, The University of Hong Kong, Pok Fu Lam, Hong Kong.
Department of Computer Science, The University of Hong Kong, Pok Fu Lam, Hong Kong.
Sci Rep. 2017 Jul 3;7(1):4536. doi: 10.1038/s41598-017-04707-4.
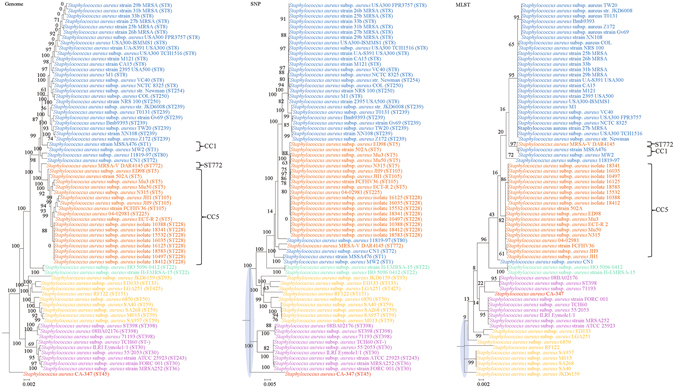
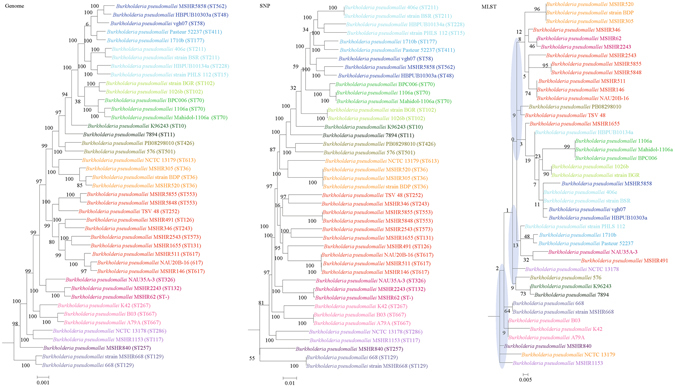
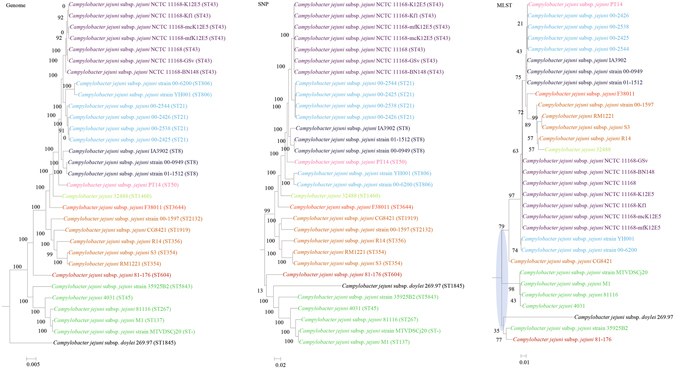
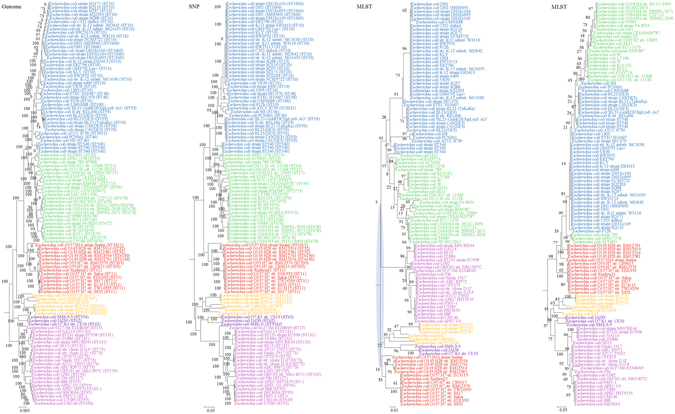
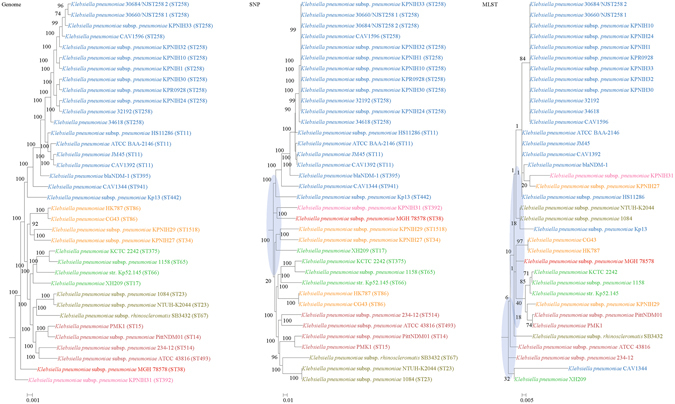
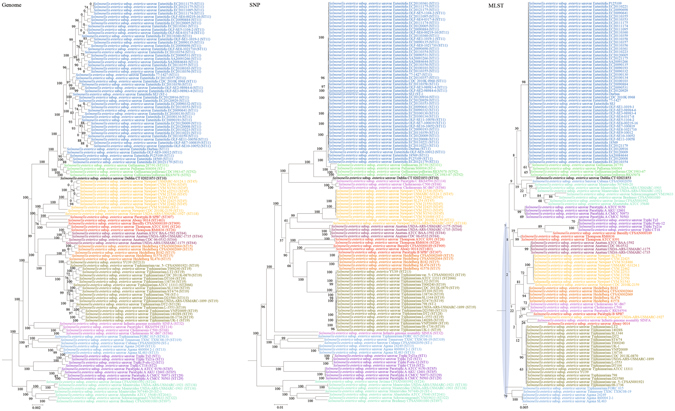
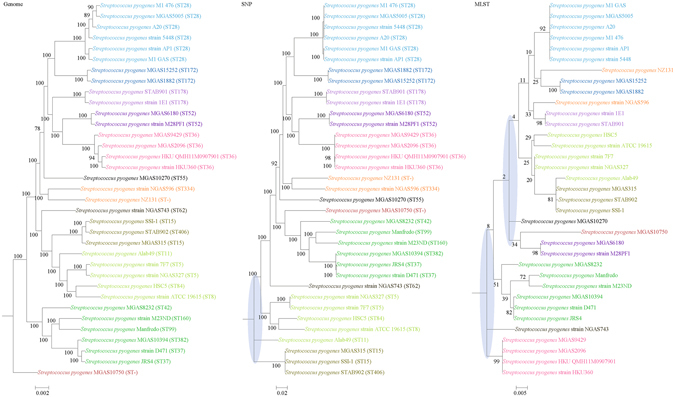
Although multilocus sequence typing (MLST) is highly discriminatory and useful for outbreak investigations and epidemiological surveillance, it has always been controversial whether clustering and phylogeny inferred from the MLST gene loci can represent the real phylogeny of bacterial strains. In this study, we compare the phylogenetic trees constructed using three approaches, (1) concatenated blocks of homologous sequence shared between the bacterial genomes, (2) genome single-nucleotide polymorphisms (SNP) profile and (3) concatenated nucleotide sequences of gene loci in the corresponding MLST schemes, for 10 bacterial species with >30 complete genome sequences available. Major differences in strain clustering at more than one position were observed between the phylogeny inferred using genome/SNP data and MLST for all 10 bacterial species. Shimodaira-Hasegawa test revealed significant difference between the topologies of the genome and MLST trees for nine of the 10 bacterial species, and significant difference between the topologies of the SNP and MLST trees were present for all 10 bacterial species. Matching Clusters and R-F Clusters metrics showed that the distances between the genome/SNP and MLST trees were larger than those between the SNP and genome trees. Phylogeny inferred from MLST failed to represent genome phylogeny with the same bacterial species.
虽然多位点序列分型(MLST)高度具有鉴别力,并且可用于暴发调查和流行病学监测,但从 MLST 基因座推断的聚类和系统发育是否能代表细菌株的真实系统发育一直存在争议。在这项研究中,我们比较了使用三种方法构建的系统发育树:(1)细菌基因组之间共享的同源序列串联块,(2)基因组单核苷酸多态性(SNP)谱和(3)相应 MLST 方案中基因座的串联核苷酸序列,用于比较 10 个具有 >30 个完整基因组序列的细菌物种。对于所有 10 个细菌物种,使用基因组/SNP 数据和 MLST 推断的菌株聚类在多个位置存在较大差异。Shimodaira-Hasegawa 检验显示,在 10 个细菌物种中的 9 个中,基因组和 MLST 树的拓扑结构存在显著差异,在所有 10 个细菌物种中,SNP 和 MLST 树的拓扑结构存在显著差异。匹配聚类和 R-F 聚类指标显示,基因组/SNP 树和 MLST 树之间的距离大于 SNP 和基因组树之间的距离。MLST 推断的系统发育未能以相同的细菌物种代表基因组系统发育。