He Maozhang, Fang Shaoming, Huang Xiaochang, Zhao Yuanzhang, Ke Shanlin, Yang Hui, Li Zhuojun, Gao Jun, Chen Congying, Huang Lusheng
State Key Laboratory of Pig Genetic Improvement and Production Technology, Jiangxi Agricultural University Nanchang, China.
Front Microbiol. 2016 Dec 23;7:2108. doi: 10.3389/fmicb.2016.02108. eCollection 2016.
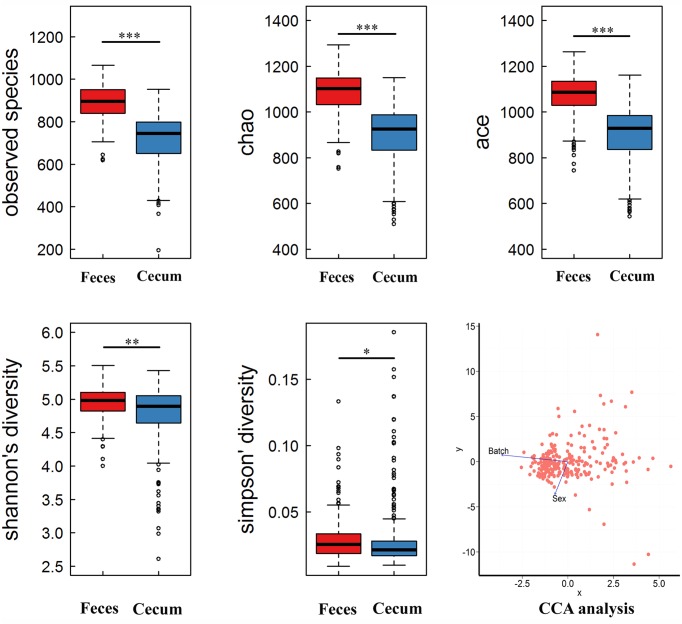
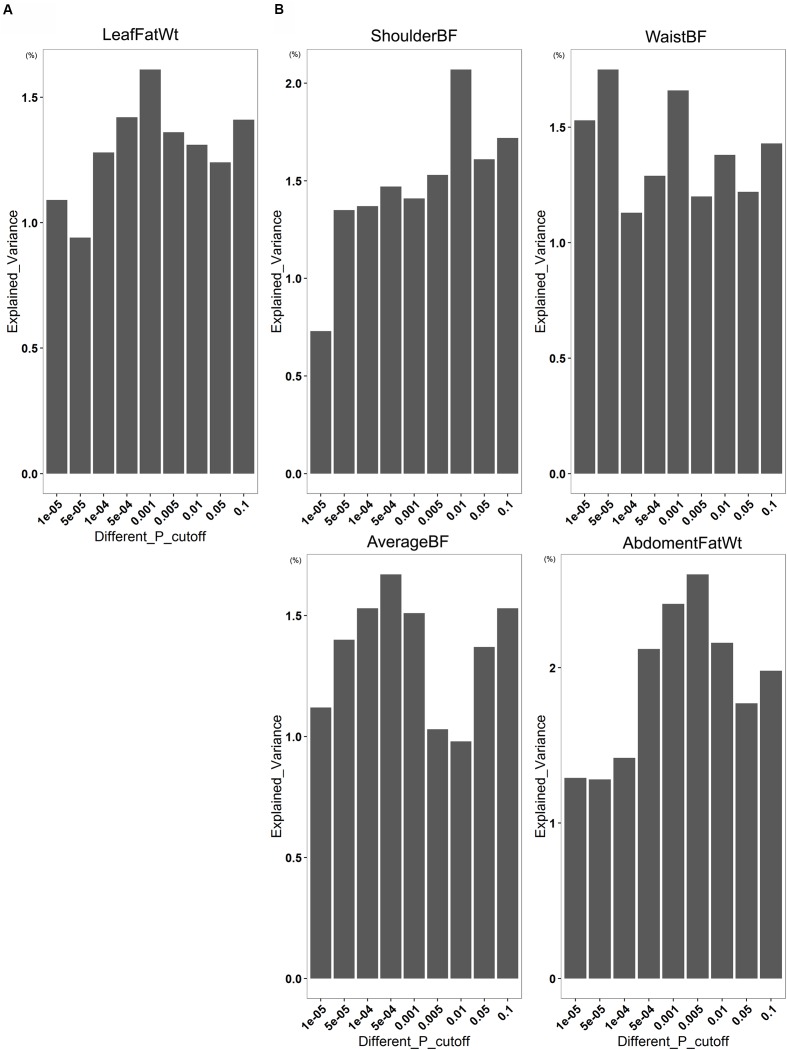
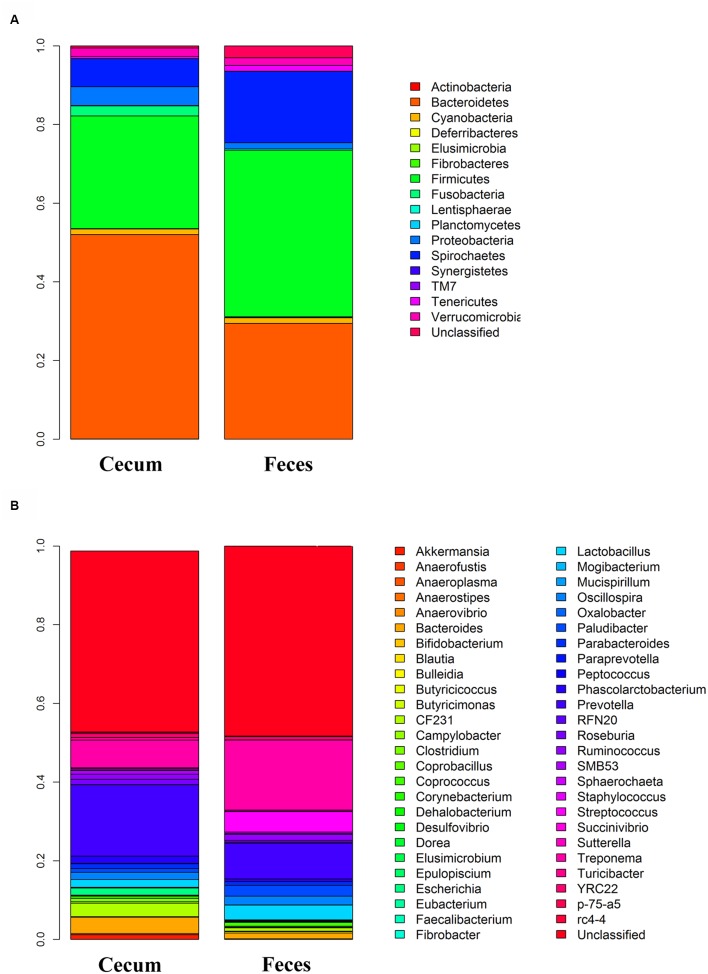
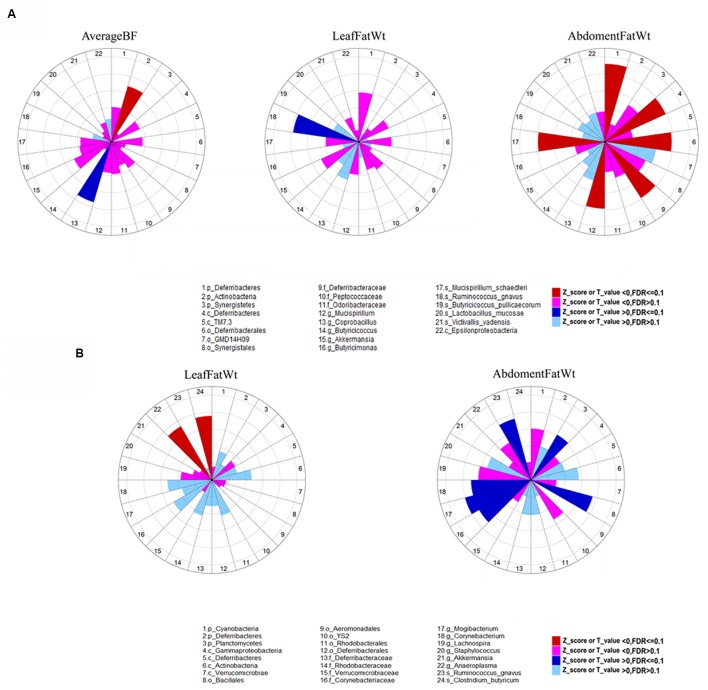
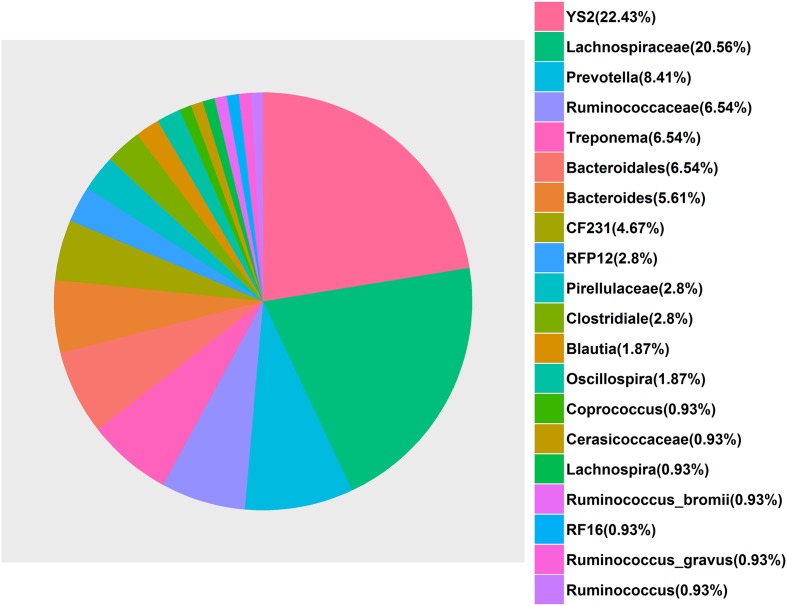
Microbial community in gastrointestinal tract participates in the development of the obesity as well as quite a few metabolic diseases in human. However, there are few studies about the relationship between gut microbiota and porcine fatness. Here, we used high-throughput sequencing to perform 16S rRNA gene analysis in 256 cecum luminal samples from pigs and 244 stools from pigs, and adopted a two-part model statistical method to evaluate the association of gut microbes with porcine fatness. As the results, we identified a total of 6 and 108 operational taxonomic units (OTUs), and 9 and 10 bacterial taxa which showed significant associations with fatness traits in the stool and cecum samples, respectively. Cross-validation analysis indicated that gut microbiome showed the largest effect on abdominal adipose by explaining 2.73% phenotypic variance of abdominal fat weight. Significantly more fatness-associated OTUs were identified in the cecum samples than that in the stools, suggesting that cecum luminal samples were better used for identification of fatness-associated microbes than stools. The fatness-associated OTUs were mainly annotated to , , , , and . These microbes have been reported to produce short-chain fatty acids by fermenting dietary indigested polysaccharide and pectin. The short-chain fatty acids can regulate host body energy homeostasis, protect host from inflammation and inhibit fat mass development. Our findings suggested that the gut microbiome may be an important factor modulating fatness in pigs.
胃肠道中的微生物群落参与了人类肥胖以及不少代谢疾病的发展。然而,关于肠道微生物群与猪肥胖之间关系的研究较少。在此,我们使用高通量测序对来自猪的256份盲肠腔样本和244份粪便样本进行16S rRNA基因分析,并采用两部分模型统计方法来评估肠道微生物与猪肥胖的关联。结果,我们分别在粪便和盲肠样本中总共鉴定出6个和108个可操作分类单元(OTU),以及9个和10个与肥胖性状显著相关的细菌分类群。交叉验证分析表明,肠道微生物群对腹部脂肪的影响最大,解释了腹部脂肪重量2.73%的表型变异。在盲肠样本中鉴定出的与肥胖相关的OTU明显多于粪便样本,这表明盲肠腔样本比粪便更适合用于鉴定与肥胖相关的微生物。与肥胖相关的OTU主要注释为 、 、 、 和 。据报道,这些微生物通过发酵饮食中未消化的多糖和果胶产生短链脂肪酸。短链脂肪酸可以调节宿主身体的能量稳态,保护宿主免受炎症影响并抑制脂肪量的增加。我们的研究结果表明,肠道微生物群可能是调节猪肥胖的一个重要因素。