Zhang Li, Wu Weida, Lee Yuan-Kun, Xie Jingjing, Zhang Hongfu
State Key Laboratory of Animal Nutrition, Institute of Animal Sciences, Chinese Academy of Agricultural Sciences, Beijing, China.
Department of Microbiology and Immunology, National University of Singapore, Singapore, Singapore.
Front Microbiol. 2018 Jan 26;9:48. doi: 10.3389/fmicb.2018.00048. eCollection 2018.
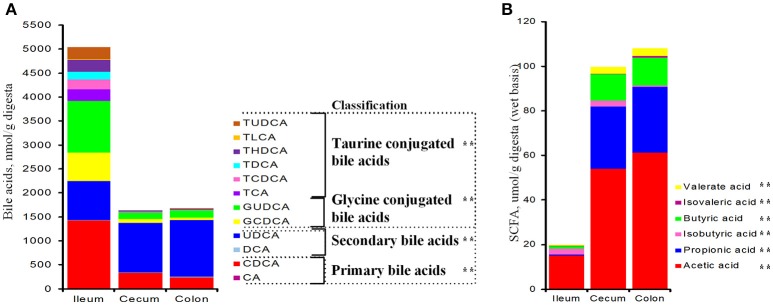
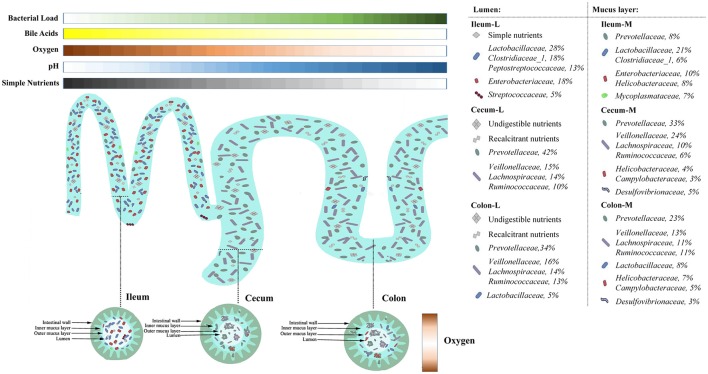
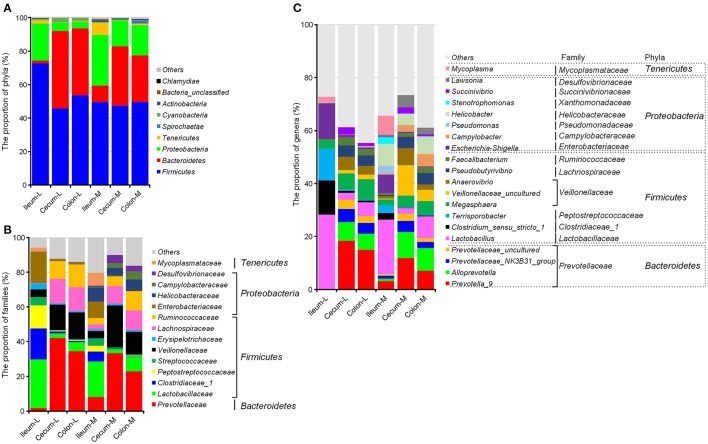
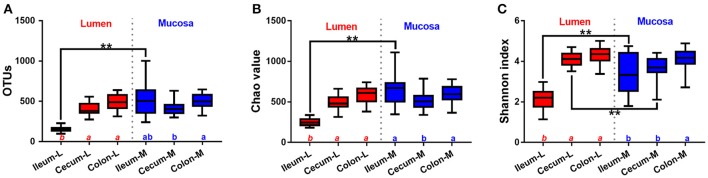
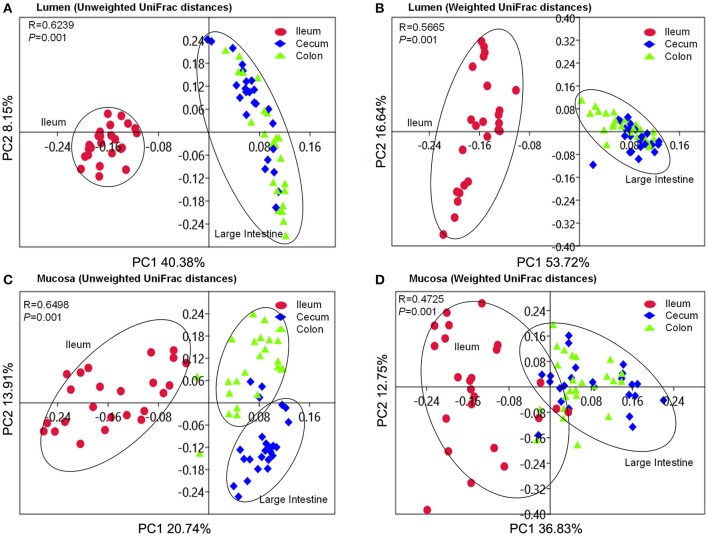
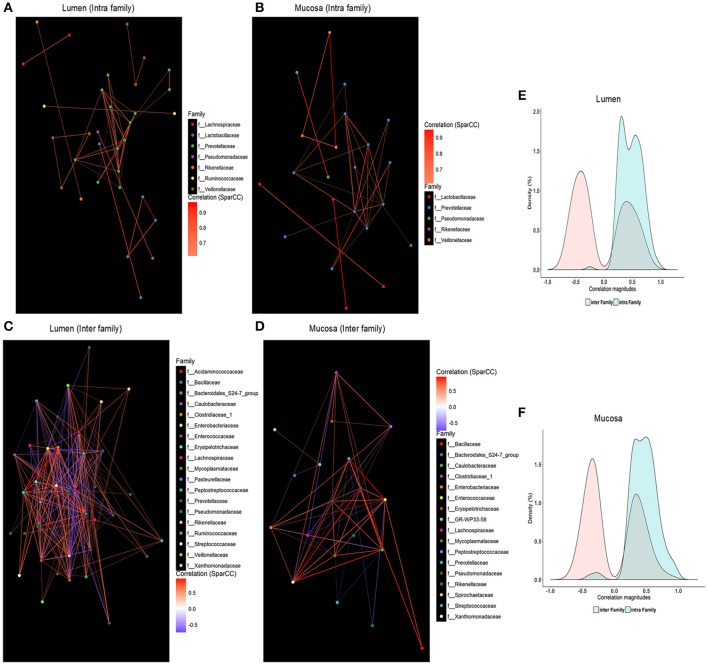
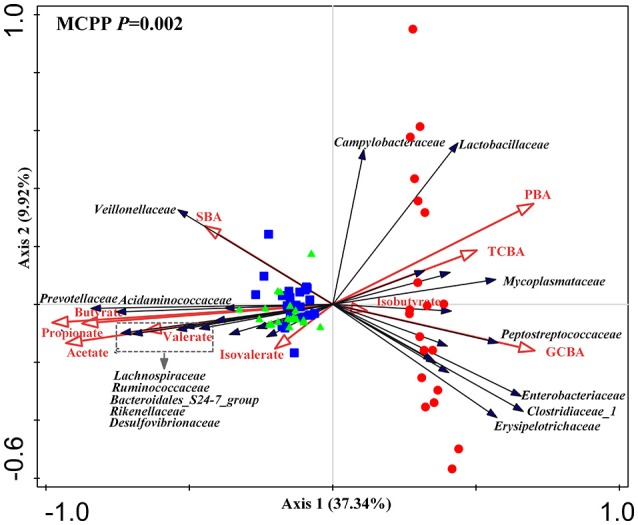
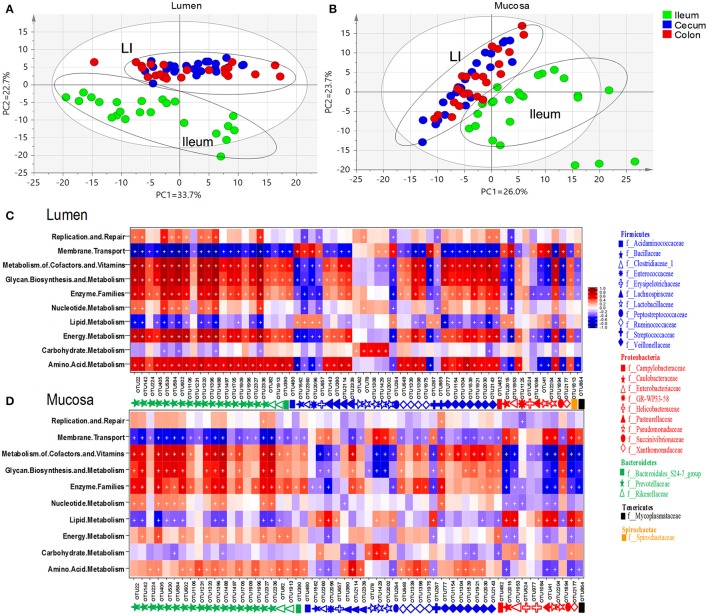
Pigs are one of the most important economic livestock. Gut microbiota is not only critical to the health but also the production efficiency of pigs. Manipulating gut microbiota relies on the full view of gut microbiome and the understanding of drive forces shaping microbial communities. 16s rDNA sequencing was used to profile microbiota along the longitudinal and radical axes to obtain the topographical map of microbiome in different intestinal compartments in young pigs. Alpha and beta-diversities revealed distinct differences in microbial compositions between the distal ileum and cecum and colon, as well as between the lumen and mucosa. and dominated in the ileum, constituting 95 and 80% of the luminal and mucosa-attached microbiome. Transitioning from the small intestine to the large intestine, luminal increased from 1.69 to 45.98% in the cecum and 40.09% in the colon, while mucosal raised from 9 to 35.36% and 27.96%. Concurrently, luminal and and mucosal-attached remarkably decreased. By co-occurrence network analyses, and were recognized as the central nodes of luminal microbial network, and and were identified as mucosal central nodes. Co-abundance was uncovered among , and in the luminal and mucosal microbiome, while opportunistic pathogens from γ- in the mucosa. Strong co-exclusion was shown between with -centered microbial groups in the lumen. Redundancy analysis found bile acids and short chain fatty acids explained 37.1 and 41% of variations in the luminal microbial composition, respectively. Primary bile acid, taurine- and glycine- conjugated bile acids were positively correlated with , whereas secondary bile acids, acetate, propionate, butyrate, and valerate were positively correlated with . Functional analyses demonstrated that , and were positively correlated with gene functions related to amino acids, energy, cofactors and vitamins metabolism, which are indispensable for the hosts. These results suggested site specific colonization and co-occurrence of swine gut microbiome closely relate to the microenvironment in each niche. Interactions of core gut microbiome greatly contributed to metabolism and/or immunity in the swine intestine.
猪是最重要的经济家畜之一。肠道微生物群不仅对猪的健康至关重要,而且对其生产效率也很关键。调控肠道微生物群依赖于对肠道微生物组的全面了解以及对塑造微生物群落驱动力的认识。利用16s rDNA测序对幼猪不同肠道区域沿纵向和径向轴的微生物群进行分析,以获得微生物组的地形图。α和β多样性揭示了回肠末端、盲肠和结肠之间以及肠腔和黏膜之间微生物组成的明显差异。在回肠中,[具体微生物名称1]和[具体微生物名称2]占主导地位,分别占肠腔和黏膜附着微生物组的95%和80%。从小肠过渡到大肠,肠腔中的[具体微生物名称1]在盲肠中从1.69%增加到45.98%,在结肠中增加到40.09%,而黏膜中的[具体微生物名称1]从9%增加到35.36%和27.96%。同时,肠腔中的[具体微生物名称3]、[具体微生物名称4]以及黏膜附着的[具体微生物名称5]显著减少。通过共现网络分析,[具体微生物名称1]和[具体微生物名称2]被认为是肠腔微生物网络的中心节点,[具体微生物名称6]和[具体微生物名称7]被确定为黏膜中心节点。在肠腔和黏膜微生物组中,[具体微生物名称8]、[具体微生物名称9]和[具体微生物名称10]之间发现了共丰度,而黏膜中来自γ-的机会致病菌存在。在肠腔中,[具体微生物名称11]与以[具体微生物名称12]为中心的微生物群之间表现出强烈的共排斥。冗余分析发现胆汁酸和短链脂肪酸分别解释了肠腔微生物组成变化的37.1%和41%。初级胆汁酸、牛磺酸和甘氨酸结合胆汁酸与[具体微生物名称1]呈正相关,而次级胆汁酸、乙酸、丙酸、丁酸和戊酸与[具体微生物名称2]呈正相关。功能分析表明,[具体微生物名称1]、[具体微生物名称2]和[具体微生物名称3]与宿主必需的氨基酸、能量、辅因子和维生素代谢相关的基因功能呈正相关。这些结果表明,猪肠道微生物组的位点特异性定殖和共现与每个生态位的微环境密切相关。核心肠道微生物群的相互作用对猪肠道的代谢和/或免疫有很大贡献。