Wang Rong, Xu Sheng, Wang Ning, Xia Bing, Jiang Yumei, Wang Ren
Institute of Botany, Jiangsu Province and Chinese Academy of SciencesNanjing, China; The Jiangsu Provincial Platform for Conservation and Utilization of Agricultural GermplasmNanjing, China.
Key Laboratory of Biology and Genetic Improvement of Soybean, National Center for Soybean Improvement, Ministry of Agriculture, Nanjing Agricultural University Nanjing, China.
Front Plant Sci. 2017 Jan 5;7:1971. doi: 10.3389/fpls.2016.01971. eCollection 2016.
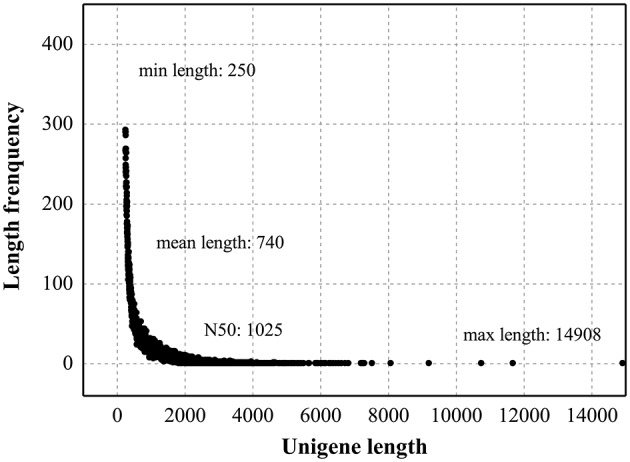
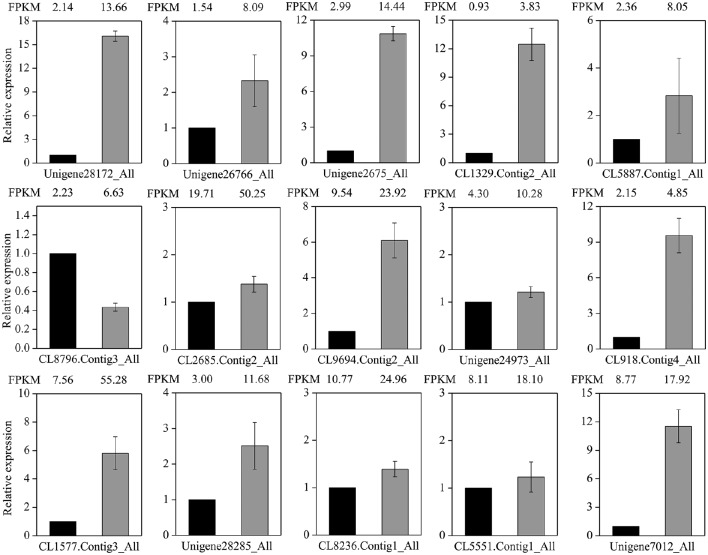
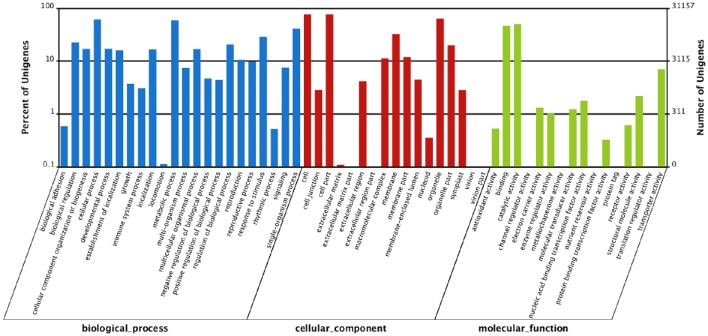
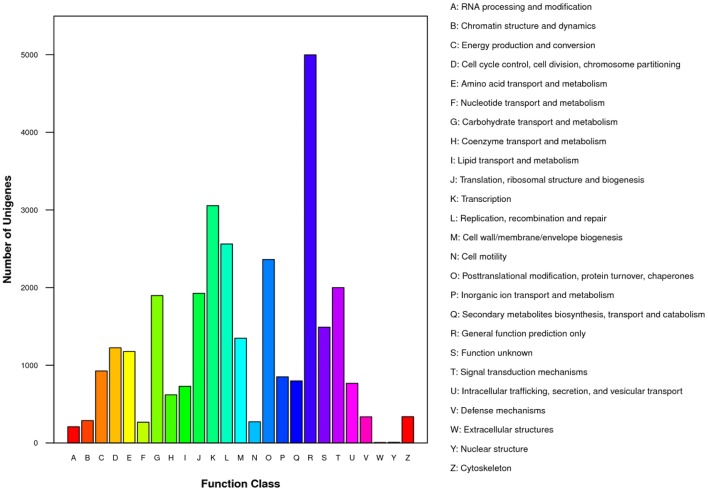
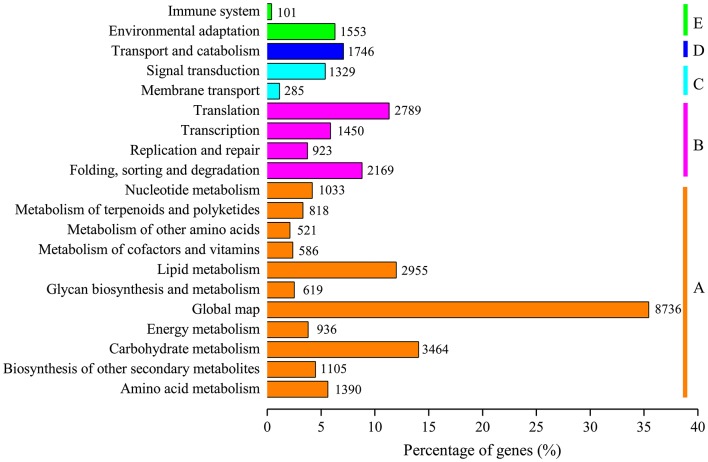
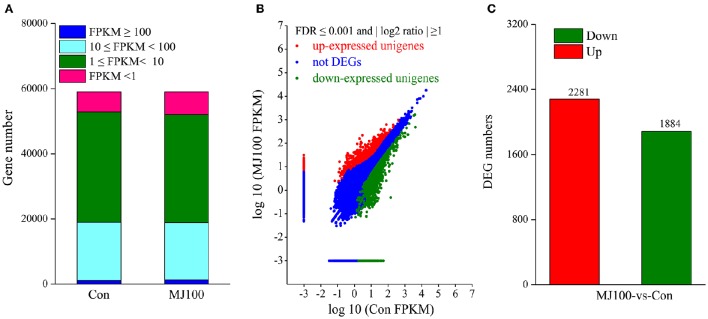
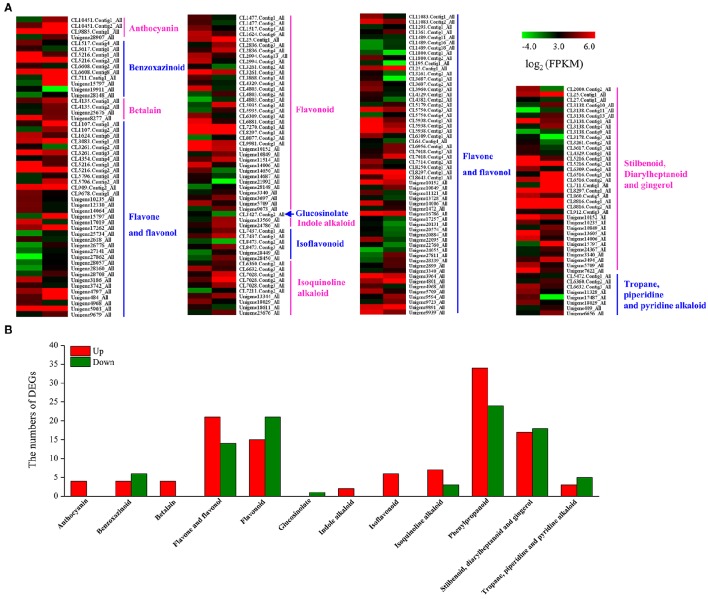
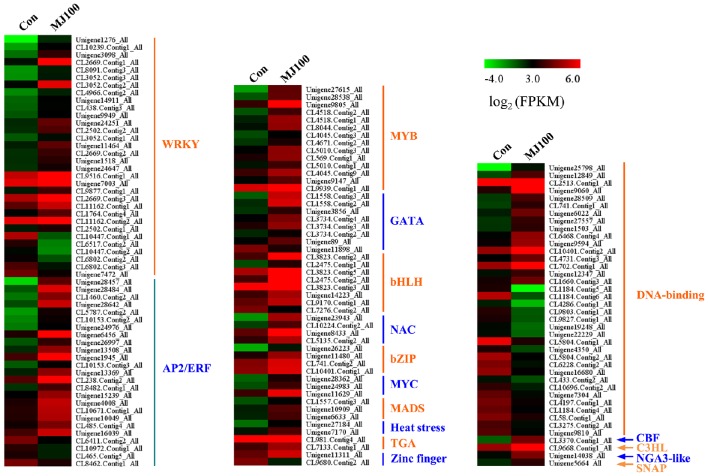
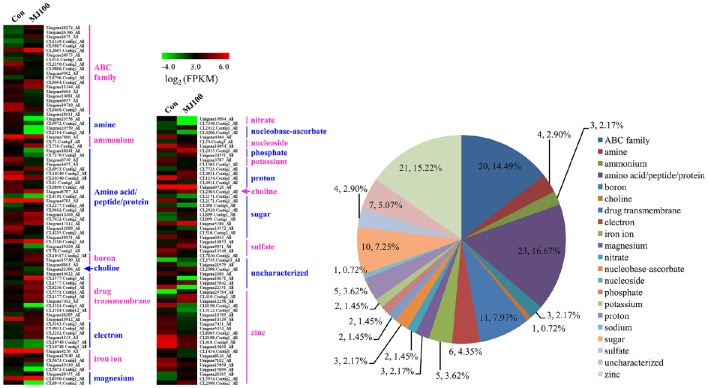
, a medicinal species of the Amaryllidaceae family, is used in the practice of traditional Chinese medicine (TCM) because of its broad pharmacological activities of Amaryllidaceae alkaloids. Despite the officinal and economic importance of species, the secondary mechanism for this species is relatively deficient. In this study, we attempted to characterize the transcriptome profiling of seedlings with the methyl jasmonate (MeJA) treatment to uncover the molecular mechanisms regulating plant secondary metabolite pathway. By using short reads sequencing technology (Illumina), two sequencing cDNA libraries prepared from control (Con) and 100 μM MeJA-treated (MJ100) samples were sequenced. A total of 26,809,842 and 25,874,478 clean reads in the Con and MJ100 libraries, respectively, were obtained and assembled into 59,643 unigenes. Among them, 41,585 (69.72%) unigenes were annotated by basic local alignment search tool similarity searches against public sequence databases. These included 55 Gene Ontology (GO) terms, 128 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and 25 Clusters of Orthologous Groups (COG) families. Additionally, 4,175 differentially expressed genes (DEGs; false discovery rate ≤ 0.001 and |log Ratio| ≥ 1) with 2,291 up-regulated and 1,884 down-regulated, were found to be affected significantly under MeJA treatment. Subsequently, the DEGs encoding key enzymes involving in the secondary metabolite biosynthetic pathways, transcription factors, and transporter proteins were also analyzed and summarized. Meanwhile, we confirmed the altered expression levels of the unigenes that encode transporters and transcription factors using quantitative real-time PCR (qRT-PCR). With this transcriptome sequencing, future genetic and genomics studies related to the molecular mechanisms associated with the chemical composition of may be improved. Additionally, the genes involved in the enrichment of secondary metabolite biosynthesis-related pathways could enhance the potential applications of .
[物种名称]是石蒜科的一种药用植物,由于石蒜科生物碱具有广泛的药理活性,因此在传统中医实践中被使用。尽管该物种具有药用和经济重要性,但其次生代谢机制相对不足。在本研究中,我们试图通过茉莉酸甲酯(MeJA)处理来表征[物种名称]幼苗的转录组图谱,以揭示调节植物次生代谢产物途径的分子机制。通过使用短读长测序技术(Illumina),对从对照(Con)和100μM MeJA处理(MJ100)样品中制备的两个测序cDNA文库进行了测序。在Con文库和MJ100文库中分别获得了总共26,809,842和25,874,478条 clean reads,并组装成59,643个单基因。其中,通过基本局部比对搜索工具与公共序列数据库进行相似性搜索,对41,585个(69.72%)单基因进行了注释。这些包括55个基因本体论(GO)术语、128条京都基因与基因组百科全书(KEGG)途径和25个直系同源群(COG)家族。此外,发现有4,175个差异表达基因(DEGs;错误发现率≤0.001且|log比值|≥1),其中2,291个上调,1,884个下调,在MeJA处理下受到显著影响。随后,还对编码参与次生代谢产物生物合成途径的关键酶、转录因子和转运蛋白的DEGs进行了分析和总结。同时,我们使用定量实时PCR(qRT-PCR)确认了编码转运蛋白和转录因子的单基因的表达水平变化。通过这种转录组测序,未来与[物种名称]化学成分相关的分子机制的遗传和基因组学研究可能会得到改善。此外,参与次生代谢产物生物合成相关途径富集的基因可以增强[物种名称]的潜在应用。